INTRODUCTION
Recently, the prevalence of a variety of immune disorders has been gradually increasing in the developed countries. One hypothesis for this phenomenon was lack of infectious diseases, including helminthic parasite infections [1]. There are many reports about the negative relationship between infectious diseases and immunological diseases, including asthma, rheumatism, allergic diseases, cardiovascular disease, Type 1 diabetes mellitus, multiple sclerosis, and inflammatory bowel diseases (IBD) [2-7].
IBD may derive from an abnormal host immune responses to normal commensal enteric bacteria, leading to a loss of immune tolerance. Although the causative mechanisms of the IBD have yet to be thoroughly established, epidemiological and laboratory works suggest that environmental and genetic factors are crucial to their pathogenesis [8,9]. Recently, new therapeutic trials for IBD have been conducted using either parasite infections or parasite-derived material treatment. Summers et al. [10] demonstrated that Trichuris suis could be an effective treatment for active Crohn's disease. T. suis has also proven to be effective in ulcerative colitis in a randomized trial conducted by the same group. In addition, experimental animal studies have revealed that infections with the helminths, Schistosoma mansoni, Hymenolepis diminuta, and Trichinella spiralis ameliorate intestinal colitis in animal models [11-13]. Many researchers have already isolated several immune down-regulatory molecules from parasites, and these molecules have been identified as mammal cytokine homologues, protease inhibitors (cystatin and serpin), abundant larval transcript antigens, and venom allergen-like proteins [14-17].
Cystatins are inhibitors of reversible cystein proteases and consist of 3 families; stefins (cystatins A and B, or Type I; MW 11 kDa), cystatins (Cystatin C, E, and S, or Type II; MW 13-14 kDa), and kininogens (Type III; 88-114 kDa) [18]. Their functions in the immune system are to modulate cathepsin activities and antigen presentation. Cystatins, type II cystatins in particular, have been shown to be important immuno-modulatory proteins, involved in inflammatory processes, antigen presentation, and lymphocyte activation [19,20]. In addition, cystatins isolated from nematode parasites can lead to drastic changes in the antigen processing and presentation of antigen presenting cells (APCs) of the host [21-24]. Especially, the cystatin (AvCystatin) isolated from Acanthocheilonema vitae could regulate expression of IL-10 in the host macrophages. This up-regulation reduced both the allergic response and the intestinal inflammation [25].
Although most of the stefins were expressed intracellularly, some stefins have been recently isolated from parasite excretory-secretory proteins. Therefore, parasite stefins might also have immunomodulatory functions, in a manner similar to nematode cystatins [26]. Previously, Kang et al. [27] isolated a stefin (CsStefin-1) from the liver fluke Clonorchis sinensis. CsStefin-1 was shown to be an active cysteine protease inhibitor; it was able to inhibit the protease activity of cathepsin Fs from C. sinensis (CsCFs), as well as human cathepsin B, human cathepsin L, and papain [27]. The antibodies against CsStefin-1 were detected in the sera of rats 14 weeks after an experimental infection with C. sinensis [27]. This result indicates that the protein could be released outside of the parasite, where it possiblly protects the parasite and host's biliary epithelial cells from the strong protease activity possessed by the CsCFs. Additional extracellular functions, including the modulation of the host immune response by the CsStefin-1, are still have to be determined. Moreover, Jeong et al. [28] suggested that the C. sinensis-derived total protein could attenuate the airway inflammation in the mouse asthma model by inducing regulatory T cells (CD4+CD25+Foxp3+T, Treg), showing that C. sinensis might act as a host immune modulator.
In this study, to investigate whether CsStefin-1 might be a new host immune modulator, mice with experimentally-induced inflammation were treated with CsStefin-1, and the inflammation response in mice was evaluated. In addition, the mechanisms related to their anti-inflammation ability were characterized.
MATERIALS AND METHODS
Preparation of the rCsStefin-1 protein secreted from C. sinensis
In a previous study, the cystatin (CsStefin-1) of C. sinensis was cloned into pQE9 (Qiagen Science, Valencia, California, USA). Recombinant CsStefin-1 (rCsStefin-1) was expressed and purified on the basis of a previously published report [27]. Purified rCsStefin-1 was desalted by HiTrap™ desalting column (Amersham Biosciences, Little Calfont, UK). Lipopolysacharide (LPS) was depleted using the Detoxi-gel endotoxin removing column (Pierce, Rockford, Illinois, USA).
Induction and assessment of colitis
Female C57BL/6 mice aged 6 weeks were purchased from Samtako Co. (Gyeonggi-do, Korea). This study included 3 groups; non-treated group (control), only colitis induced group, and rCsStefin-1 treated group (after induction of colitis). There were 5 mice in each group. From day 0 to day 4, mice were administered 3% (wt/vol) dextran sulfate sodium (DSS) in drinking water (molecular weight, approximately 40,000) (MP Biomedicals, LLC, France). From day 4 to day 8, mice were treated 4 times with i.p. injections of rCsStefin-1 with a dose of 2 µg. Mice were weighed and visually observed for rectal bleeding and diarrhea every day, starting with day 0. The disease activity index (DAI) was used to assess the grade of colitis. The score ranges from 1 to 4, and represents the sum of the scores for body weight loss, stool consistency, and rectal bleeding divided by 3 (Table 1).
Histological evaluation of colitis
The colon tissue samples were isolated from mice, and washed with PBS. Then, the tissue samples were fixed with 4% paraformaldehyde, embedded in paraffin, mounted on glass slides, and stained with hematoxylin and eosin. For histological evaluation of the tissue damage, areas of inflammatory lesions were microscopically evaluated and scored, as shown in Table 2.
Cytokine production
After mice were killed, lymphocytes from their spleen and mesenteric lymph nodes (MLNs) were isolated to investigate the incidence of specific cytokine secreting lymphocytes. Spleens and MLNs were treated with ACK hypotonic lysis solution (Sigma-Aldrich, Saint Louise, Missouri, USA) for 2 min at the room temperature, for RBC lysis. After RBCs were lysed, the remaining cells were filtered through 100 µm meshes (Small Parts Inc., Seattle, Washington, USA), and were plated in 48 well plates, as 5×106 cells/ml in RPMI 1640 with 10% fetal bovine serum (FBS) and penicillin/streptomycin. For the CD3 stimulation experiments, 0.5 µg/ml of CD3 antibody (eBioscience, San Diego, California, USA) was added to the cell-plated well. Plated cells were then incubated for 72 hr at 37℃ in air containing 5% CO2. After incubation, culture media were harvested and stored at -20℃.
ELISA
ELISA tests were performed to measure IL-10, TGF-β, TNF-α, IL-1β, and IFN-γ in culture supernatants with the ELISA kit (eBioscience). Each assay was carried out according to the manufacturer's recommended protocols.
Realtime PCR
The colon tissue was immersed in 1 ml QIAzol (Qiagen Science), and homogenized with a tissue homogenizer. RNA extraction was performed following the manufacturer's protocols (Qiagen Science), transcribing 2 µg RNA with moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Madison, Wisconsin, USA). IL-10 RNA levels were measured by real-time PCR using the iCycler™ (Bio-Rad Laboratories, Inc., Hercules, California, USA) real-time PCR equipment and GAPDH were used as reference genes. Primers were produced according to a previous report [29]. PCR amplification was carried out in 20 µl solution, containing 2 µl cDNA, 0.2 µM primers, and the SYBR Premix Ex Taq™ (TAKARA Bio Inc., Otsu, Shiga, Japan). Expression levels were estimated using the iCycler iQ™ multi-color real-time PCR detection system (Bio-Rad). The relative expression of the gene was then calculated as the ratio to a housekeeping gene, using the gene-x program (Bio-Rad).
Signal pathway
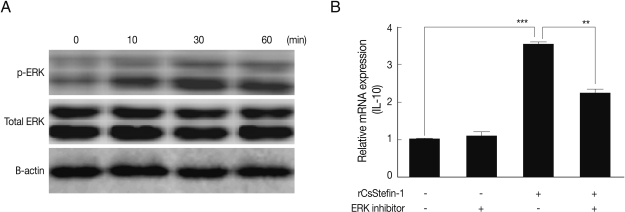
A mouse colon epithelial cell line (CT-26 cells) was maintained in Dubecco's modified Eagle's medium (DMEM) (Hyclone, Road Logan, Utah, USA), supplemented with 10% heat-inactivated FBS (Hyclone), 100 units/ml penicillin and 100 µg/ml streptomycin. A 1-µg/ml rCsStefin-1 protein was added to cells for 0, 10, 30, and 60 min. CT-26 cells were harvested by scrapper (Bio-Rad) and washed twice with cold PBS. The total protein of CT-26 cells was extracted using the PRO-PREP protein extraction solution (Intron Biotechnology, Sungnam, Korea). The extraction procedures were carried out according to the manufacturer's recommended protocols. The protein concentration was determined by the Bradford method (Bio-Rad). These samples were separated into 30 µg fragments by SDS-PAGE on 10% acrylamide gel, which were then transferred onto nitrocellulose membranes (Amersham Biosciences), blocked with 5% skim milk in TBS containing 0.1% triton x-100 (TBST) for 1 hr at room temperature, and the membrane was washed 5 times with TBST. To detect the proteins, the membrane was incubated with appropriate antibodies. These were anti-mouse-pERK monoclonal antibodies (mAb) (Santa Cruz Bio Inc., Santa Cruz, California, USA), anti-mouse-p38 mAb (Cell Signaling Technology, Inc., Beverly, Massachustts, USA), and anti-mouse-SAPK/JNK mAb (Cell Signaling Technology). As the secondary antibody, the anti-mouse IgG-HRP conjugate was purchased from Sigma. First antibodies were used at a 1: 1,000 dilution in 1% BSA TBST. HRP conjugated secondary antibodies were used at a 1:2,000 dilution. ECL substrate (Amersham Biosciences, Uppsala, Sweden) was used for detection.
Flow cytometry analysis
After mice were killed, immune cells from their spleen and MLNs were isolated to investigate IL-10 secreting cell recruitment. At first, to evaluate the recruitment of Treg cells induced by rCsStefin-1 treatment, isolated cells from the spleens and MLNs were subjected to CD4, CD25, and Foxp3 staining using FITC-labeled anti-mouse CD4, APC-labeled anti-mouse CD25, and Pacific Blue-labeled anti-mouse Foxp3 antibodies (eBioscience). The cell surfaces were then stained with anti-mouse CD4 and anti-mouse CD25 antibody, in accordance with the manufacturer's recommended protocols. After surface staining, the cells were treated for 30 min with permeabilization buffer at 4℃ following Foxp3 staining. After treatment with permeabilization buffer, the cells were stained with anti-mouse Foxp3 antibodies.
In addition, to determine the mechanisms of elevation of IL-10 level by the rCsStefin-1 treatment, the immune cells were subjected to IL-10 staining by using the anti-mouse CD4-FITC labeled antibody, anti-F4/80-PerCP-blue labeled antibody, and PE-labeled IL-10 antibody (eBioscience). After staining with the surface marker, the anti-F4/80 antibodies and anti-CD4 antibodies, the cells were treated with permeabilization buffer for 30 min at 4℃, to follow the IL-10 staining. The cells were stained with the IL-10 antibody. Flow cytometry was performed and analyzed on the FACSCanto™ ‖ (BD) with the FACS Diva™ 6.0 software.
RESULTS
Amelioration of DSS-induced intestinal inflammation after treatment with recombinant parasite cystatins
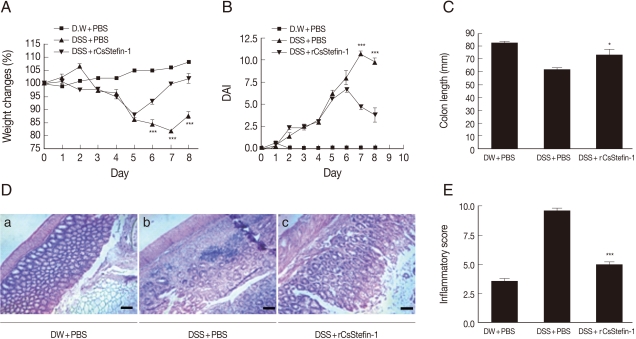
To investigate whether the recombinant parasite cystatin (rCsStefin-1) could regulate inflammation, we evaluated the intestinal inflammation after treatment with recombinant rCsStefin-1 of the DSS-induced intestinal inflammation in the mouse model of IBD. We observed a loss of body weight in the DSS-treated group, but the degree of weight loss in rCsStefin-1 treated mice was lower than thqat in DSS only-treated mice (Fig. 1A). We evaluated the severity of colitis using the percentage of weight loss, changes in the stool states, including consistency, and blood presence in the stool. These measurements have been employed as visible parameters for the degree of inflammation in the DSS-induced colitis, and expressed as DAI value. The DAI value was increased on day 2 and reduced on day 7 in DSS only-treated mice, but the DAI value in rCsStefin-1-treated mice was reduced on day 6. The DAI value was significantly lower in rCsStefin-1-treated mice than those in DSS only-treated mice on days 7 and 8 (Fig. 1B). In cases of persistent diarrhea, the colon may be shortened, along with severe inflammation of the large intestine [30]. After the mice were sacrificed, we observed that the colon of DSS only-treated mice was shorter than the colon of rCsStefin-1 treated mice (Fig. 1C). These results indicated that DSS treatment might induce severe inflammatory responses in the colon, but these responses seemed to be inhibited by the rCsStefin-1 treatment.
Fig. 1D showed histological changes in the colon after DSS and/or rCsStefin-1 treatment. In the histological examination, the colon of control mice maintained structurally intact epithelium and submucosal layers (Fig. 1D-a). However, the submucosal layers of the colon in DSS-treated mice showed a variety of inflammatory cell infiltrations and ulcerative mucosa, as well as destructed crypt cells in the epithelium (Fig. 1D-b). In contrast, the majority of the epithelium was intact and the crypt cells and ulcerative lesions were only rarely detected in the colon of rCsStefin-1-treated mice (Fig. 1D-c). The tissue histopathology index scores were significantly higher in DSS-treated mice than those in mice treated with rCsStefin-1 after DSS-treatment (Fig. 1E). These results demonstrated that rCsStefin-1 treatment may facilitate the repair of the ulcerations induced by DSS.
TNF-α production in MLNs was inhibited by rCsStefin-1 treatment, but IL-10 production was elevated
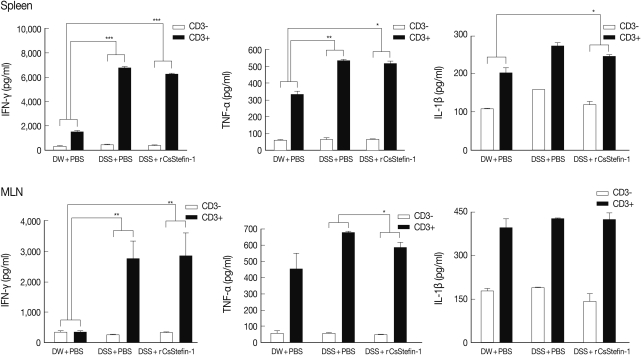
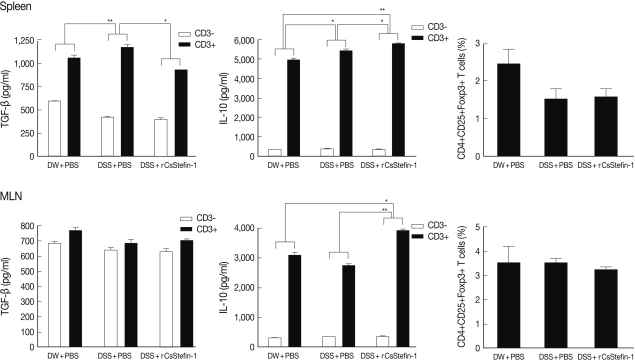
To investigate the mechanism of protection against intestinal inflammation elicited by rCsStefin-1, we evaluated IFN-γ, TNF-α, IL-1β, and regulatory cytokine levels of lymphocytes isolated from MLNs and spleen of mice with DSS-induced intestinal inflammation after rCsStefin-1 treatment (Fig. 2). The secretion levels of IFN-γ, TNF-α, and IL-1β of splenocytes were significantly increased after the DSS treatment, but those levels were not significantly reduced after rCsStefin-1 treatment (Fig. 2). However, the levels of TNF-α in MLNs of the rCsStefin-1-treated mice were significantly reduced. The levels of IFN-γ and IL-1β in MLNs were not changed by rCsStefin-1 treatment. To characterize the inhibitory mechanisms of the inflammatory related cytokines production, we evaluated the Treg cell recruitment and the Treg related cytokine production. The results showed that IL-10 level was significantly increased by rCsStefin-1 treatment in both the spleen and MLNs. However, the level of TGF-β was not changed (Fig. 3). The population of the Treg cells was not changed by the rCsStefin-1 treatment both in the spleen and MLNs (Fig. 3).
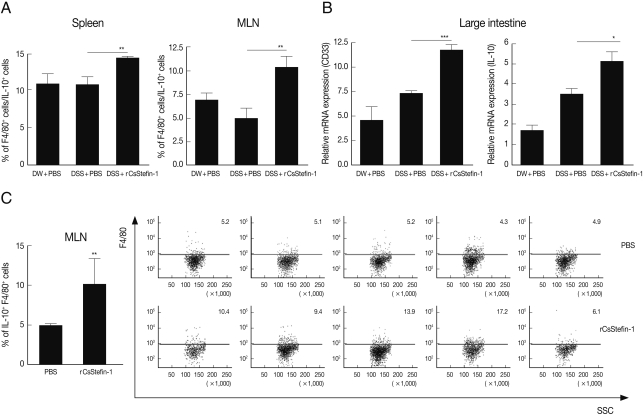
IL-10 secreting macrophage was increased by rCsStefin-1 treatement
To know increased IL-10 secreting cell subtype, we analyzed IL-10+, CD4+, and F4/80+ (macrophage-specific surface marker) cell population. In addition, we evaluated another macrophage specific gene (CD33) expression level in the large intestinal tissue after DSS and rCsStefin treatments [31,32]. We found IL-10+ CD4+ cell population in the spleen and MLNs was not changed by CsStefin-1 treatment on DSS treated mice (data not shown). Although the population of IL-10 secreting T cells was not changed, the IL-10+F4/80+ macrophage cell population was significantly increased both in the spleen and MLNs by CsStefin-1 treatment on DSS treated mice (Fig. 4A). In addition, we found CD33 and IL-10 gene expression level in large intestine of rCsStefin-1 treated mice was significantly higher than those of only DSS treated mice (Fig. 4B). We found IL-10+F4/80+ cells production in the MLNs could be significantly increased by rCsStefin-1 treatment in normal mice (Fig. 4C).
IL-10 was produced by rCsStefin-1 treatment via the ERK pathway
We found that the IL-10 expression level in the large intestinal tissue was significantly increased by rCsStefin-1 treatment (Fig. 4B). We also conducted western blotting to evaluate the MAPKs phosphorylated by rCsStefin-1 in intestinal epithelial cells. As a result, at 10, 30, and 60 min after rCsStefin-1 treatment, the phosphorylation of both ERK were induced, at a concentration of 1 µg/ml (Fig. 5A), whereas rCsStefin-1 did not activate the phosphorylation of p38 and SAPK/JNK in CT-26 cells (data not shown). To further evaluate the role of MAPK in IL-10 production, we conducted real-time PCR. We employed inhibitors of specific MAPKs, and examined the effects of the PD98059 (ERK1/2 inhibitor) on the CT-26. The up-regulation of IL-10 induced by the rCsStefin-1 in the CT-26 cells was prevented by pretreatment with PD98059 (Fig. 5B).
DISCUSSION
In previous report, Kang et al. [27] identified and characterized the type I cystatin (CsStefin-1) isolated from the liver fluke C. sinensis. Type I cystatins are generally described as intracellular cystein protease inhibitors. They are also found in various body fluids in healthy humans [33,34]. In addition, the CsStefin-1 was expressed throughout various developmental stages of the parasite, from metacercaria to the adult worm [27]. The structure of the CsStefin-1 (Type I cystatin) is different from the Type 2 cystatin (i.e. the filarial cystatin). Type I cystatins have no SND motif which could inhibit either the legumain, or the asparaginyl endopeptidase. Legumain like asparaginyl endopeptidases are important for the intracellular processing of antigens for presentation with the MHC class II [35]. Although there is no evidence that the CsStefin-1 could block the antigen processing of the host or not, probably this might not occur. However, it has been shown that the CsStefin-1 had another immune modulating function, which was activation and proliferation of the IL-10 secreting macrophages.
In the IBD pathogenesis, the imbalance between proinflammatory cytokines (IL-1, IL-1β, IL-2, IL-6, IL-8, IL-12, IL-17, IL-23, TNF-α, and IFN-γ) and anti-inflammatory cytokines (IL-4, IL-10, IL-11) is also important [36]. Especially, the TNF-α is the key cytokine in the pathogenesis of IBD. Therefore, most useful therapeutic agents against IBD were antibodies against TNF-α cytokines, such as infliximab, adalimumab, and certolizumab [36,37]. Severe forms of colitis, such as the Crohn's disease refractory to conventional therapy, fistulizing Crohn's disease and chronic active ulcerative colitis, generally respond well to the anti-TNF-α therapy. However, this medication can cause serious side effects, that usually occur, careful monitoring of the therapy should be performed [38,39]. Potential side effects of anti-TNF therapy include opportunistic infections, antibody formation against anti-TNF, and risks of lymphomas in the IBD patients [37,39]. New therapeutic methods should be continually investigated. Although the overall effect was not dramatic, the rCsStefin-1 treatment inhibited the level of the TNF-α cytokines in the MLNs (Fig. 2). These results suggest that rCsStefin-1 could be considered as a new anti-TNF agent.
Inhibition of the TNF-α production might occur due to increase of anti inflammatory cytokine IL-10 production. We observed that the IL-10 production was significantly increased by the rCsStefin-1 treatment, in both the spleen and the MLNs (Fig. 3). In addition, the IL-10 mRNA level in large intestine tissue was increased on rCsStefin-1 treatment (Fig. 4B). The IL-10, which is referred to as the cytokine synthesis inhibitory factor, is an anti-inflammatory cytokine capable of inhibiting the synthesis of pro-inflammatory cytokines [40]. The IL-10 deficient mice develop IBD following colonization of the gut with particular microorganisms [41]. The helminthic parasites stimulate the production of immunoregulatory mediators, which likely play a role in the maintenance of the chronicity of infection, without any marked induction of pathology. In particular, elevated levels of IL-10 and TGF-β have been associated with both the helminth infection, and the helminth-derived molecule treatment [9,29]. The IL-10 is mainly generated by the Treg cells, and both the IL-10 and the TGF-β cytokines have been shown to induce Treg cell differentiation [42]. The Treg cells perform a crucial function in the prevention of uncontrolled inflammatory responses, including IBD [43]. However, in this study, the population of Treg cells has not been changed by the rCsStefin-1 treatment (Fig. 3). Moreover, the TGF-β cytokine, which was very important for activation and proliferation of the Treg cells, was not increased in both the spleen and the MLNs. Therefore, we thought that inhibition of intestinal inflammation after rCsStefin-1 treatment might be not due to Treg cell activation, but due to other IL-10 secreting cells. In this study, we found IL-10+F4/80+ macrophage in the spleen and the MLNs was significantly increased by rCsStefin-1 treatment in DSS treated mice (Fig. 4A). In addition, we found macrophage specific CD33 gene expression in large intestine of rCsStefin-1 treated mice after DSS treatment was significantly higher than those of DSS treated mice (Fig. 4B). These results showed that IL-10+ macrophage cell subset played a role in elevation of IL-10 by CsStefin-1 treatment. In previous reports suggested that activation and proliferation of the IL-10 secreting macrophages was also increased by other parasite cystatin treatment [24,25]. In vitro, in intestinal epithelial cell lines, we have demonstrated that rCsStefin-1 induced IL-10 gene expression via phosphorylation of the MAPK ERK1/2 and that their expression was partially blocked by ERK1/2 inhibitors (Fig. 5). Although IL-10 expression may involve signaling via the ERK1/2 MAPK pathway, there might be possibility of another pathway involved in this expression because of IL-10 expression was not completely blocked by ERK1/2 inhibitor. In a recent study, it was demonstrated that parasite cystatin (AvCystatin, Type 2 cystatin) may also trigger an ERK1/2 and p38-dependent signal transduction pathway in the mouse macrophage [24].
Parasites regulate or suppress their host immune responses to maintain their parasitism for a prolonged period; the molecules governing this process are not yet identified. In this study, we determined that rCsStefin-1 may be one of these molecules regulating the host immune; thus rCsStefin-1 could prove useful in the design of novel therapeutic intervention strategies for IBD.