INTRODUCTION
The intestinal trematode, Haplorchis taichui, is a medically important parasite infecting humans and livestock. Haplorchiasis is a major public health threat in Asia and in parts of Africa and the Americas [1-3]. H. taichui is the most frequently reported species among the minute intestinal flukes from Southeast Asia, including Thailand, Lao PDR, China, and Vietnam [3,4-7]. Mitochondrial (mt) genomes exhibit a relatively conserved suite of protein-coding sequences, but also relatively rapid rates of evolutionary change [8,9]. In recent years, complete mitochondrial DNA (mtDNA) sequences have been extensively used to infer higher level phylogenies [10,11] and also for taxonomy and population genetics at lower taxonomic levels [12-14]. To date, quite a number of complete mt genomes of metazoan species, including helminths, have been deposited in GenBank and published [15]. Information from flatworm mitochondrial genomes is strongly biased toward parasitic species of medical importance. For this reason, recent mitochondrial genome scale phylogenetic surveys have emphasized the need to collect data for the major groups of flatworms that have not been sampled [16,17]. However, most of them still remain poorly understood at the molecular level, in particular, the complete mt genomes of the species in the family Heterophyidae. Parasitic flatworm mt genomes, ranging in size usually from 13 to 14 kb but far bigger up to 24 kb sometimes, are typically circular and usually encode 36 genes, including 12 protein-coding genes, and without introns and with short intergenic regions [18]. The Digenea currently contains about 18,000 nominal species parasitizing vertebrates, and sometimes humans as the definitive host [19]. The purpose of the present study was to sequence the mt genome of H. taichui for comparison with the organization and sequence of the mt genomes of other trematodes. In addition, we wished to reconstruct the phylogenetic relationships of the family Heterophyidae within the class Trematoda using mtDNA sequences.
MATERIALS AND METHODS
Long PCR amplification and sequencing of the H. taichui mtDNA molecule
Adult H. taichui worms were obtained from naturally infected Laotian people during the activity of "Korea-Lao PDR Collaborative Project for Control of Foodborne-Trematode Infections (esp. Opisthorchiasis) in Lao PDR (2007-2011)". The specimens were washed in normal physiological saline and identified based on morphological characters (gonotyl bears 12-16 spines in H. taichui). The worms were stored in 70% ethanol prior to DNA extraction. Total genomic DNA was extracted from 200 specimens using a QIAamp tissue kit (Qiagen Inc., Valencia, California, USA), according to the manufacturer's instruction and used as the template DNA. The complete mt genome was PCR-amplified in overlapping fragments. The nucleotide sequences and relative positions of the PCR primers are shown in Fig. 1. PCR reactions were performed in a 50 µl reaction volume consisting of 10 units of EF-Taq polymerase (Solgent Co., Daejeon, Korea), 2.5 mM dNTP mixture, 2.5 mM MgCl2, 20 pmole of each primer, and 10 µg of genomic DNA in a thermocycler (Biometra Co., Goettingen, Germany) under the following conditions: 92℃ for 2 min (initial denaturation), then 92℃ for 10 sec (denaturation), 50℃ for 30 sec (annealing), and 68℃ for 2 min to 10 min (extension) for 10 cycles, followed by 92℃ for 2 min, then 92℃ for 10 sec, 48℃ for 30 sec, and 68℃ for 2 min to 10 min for 20 cycles, and a final extension at 68℃ for 8 min.
A negative control (no template) was also included for every PCR reaction to detect contamination. Each amplicon (3 µl) was examined by agarose (1%) gel electrophoresis, stained with ethidium bromide and photographed using a gel documentation system (UVItec, Cambridge, UK), excised under long-wavelength UV light, extracted using a Doc-do purification kit (Elpis Co., Daejeon, Korea), and then used as a template for sequencing reactions. The primer walking method was employed to obtain overlapping sequences for each of the amplified fragments. Cyclic sequencing from both ends of the fragments was performed with a Big-Dye, and the amplified products were subjected to electrophoresis on an ABI 3100 automated DNA sequencer.
Sequence analysis and characterization of the Haplorchis taichui mt genome
Gene annotation, genome organization, translation initiation, translation termination codons, and the boundaries between protein-coding genes of mt genomes were identified based on comparison with mt genomes of other trematodes reported previously [17,18]. Sequences were assembled manually and aligned against the complete mt genome sequences of our own Metagonimus yokogawai sequence (will be published elsewhere) and trematode parasites available in the GenBank database (http://www.ncbi.nlm.nih.gov WebGenBank) using BLAST searches. Open reading frames and gene boundaries were confirmed by comparing with M. yokogawai nucleotide sequences. The codon usage profiles of 12 protein-coding genes and their nucleotide composition were calculated using Geneious 6.1.5 (Biomatters Co., Auckland, New Zealand) program [20]. Putative secondary structures of 22 tRNA genes were identified manually by recognizing potential secondary structures and anticodon sequences.
Phylogenetic analysis
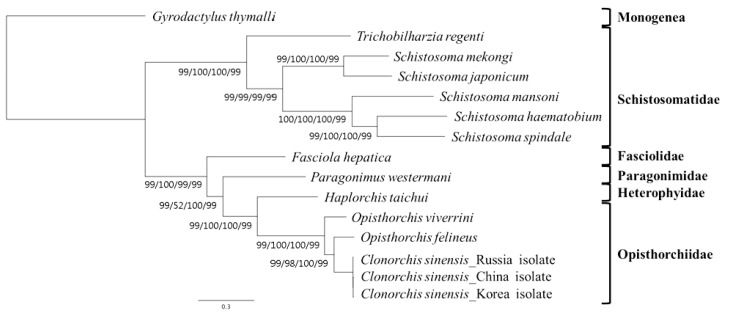
To assess the phylogenetic position of H. taichui and the utility of mt genomes in resolving the interrelationships of trematode orders, complete mitochondrial genome sequences of 15 flatworms were analyzed. The mtDNA sequences were as follows: H. taichui (KF214770), Trichobilharzia regenti (NC_009680), Schistosoma spindale (DQ157223), S. haematobium (DQ157222), S. mansoni (NC_002545), S. japonicum (NC_002544), S. mekongi (NC_002529), Fasciola hepatica (NC_002546), Paragonimus westermani (NC_002354), Opisthorchis felineus (EU921260), O. viverrini (JF739555), Clonorchis sinensis (FJ381664/JF729303/JF729304), and 1 Monogenea species (Gyrodactylus thymalli, NC_009682) as the outgroup. Each gene was translated into an amino acid sequence using the trematode mt genetic code by translation table 21 in Geneious 6.1.5, and was aligned based on its amino acid sequence using default settings. A conserved block of concatenated alignment was selected using the Gblocks program [21], (http://molevol.cmima.csic.es/castresana/Gblocks_server) for 12 protein-coding genes examined. Phylogenetic relationships among trematodes were inferred using different tree construction algorithms, i.e., maximum parsimony (MP), neighbor-joining (NJ), maximum likelihood (ML), Bayesian phylogeny (BP) [22], PAUP* [23], PhyML 3.0 [24], and MrBayes [25]. Bootstrap analysis was done using 1,000 random replications. Models of amino acid substitution were determined for each data partition independently using ModelTest supported in Geneious 6.1.5. The MP analysis was performed using the exhaustive search option. ML analysis was performed using the substitution model Le Gascuel (LG). In BP analysis, the following settings were applied: the number of cycles=1,100,000; sampling frequency=200; heated chains=4; burn-in=100,000.
RESULTS
General features of the mt genome of H. taichui
The complete mt genome of H. taichui Lao PDR isolate (GenBank accession no. KF214770) is 15,130 bp in length (Fig. 1). It encodes 36 genes; 12 protein-coding genes (cox1-3, nad1-6, nad4L, atp6, cob, and lacking atp8), 22 transfer RNA genes, and 2 ribosomal RNA genes.The relative positions and lengths of each gene are given in Table 1. An AT-rich region (1,710 bp) is located between tRNA-Glu and tRNA-Gly. All genes are transcribed in the same direction. The 2 adjacent genes, nad4L and nad4, overlap each other by 40 nt in different reading frames.
Codon usage and protein-coding genes
The mt genome of H. taichui encodes 12 protein-coding genes, identical with the situation in other trematodes. The start and termination codons of these were identified by sequence comparison with homologs in other trematodes. The ATG codon was used in 9 protein-coding genes, and the GTG codon in 3 genes (nad1, nad4, and nad5). The TAG stop codon was used in 9 genes (cox3, cob, nad4L, atp6, nad2, nad1, nad3, cox1, and nad6) and the TAA termination codon in the remaining 3 genes (nad4, cox2, and nad5). Pairwise comparisons were made among the amino acid sequences inferred from individual protein-coding genes in the H. taichui genome with those representing 12 other trematodes (Table 2). The amino acid sequence similarities in individual inferred proteins ranged from 76.8% (cox1) to 82.4% (cob) between H. taichui and O. felineus; and from 78.0% (cox1) to 80.5% (cob) between H. taichui and C. sinensis. The amino acid sequence similarities between H. taichui and S. japonicum ranged from 24.2% (cox3) to 67.9% (cox1); and from 47.1% (nad2) to 75.6% (cob) with F. hepatica (Table 2). The 12 protein-coding genes were 10,176 bp in length and composed of 43% T, 17.1% A, 28% G, and, 11.9% C, accounting for 67.3% of the full length of the genome (Table 3). All 64 codons were used (Table 4). However, some codons, such as CGC, CGA for arginine, ACA for threonine, or GCA for alanine, were very uncommon, reflecting the nucleotide composition. Several amino acids, histidine (1.6%), lysine (1.3%), and glutamine (0.7%), were rarely used. Five of the most frequently used amino acids were leucine (16.7%), valine (11.9%), serine (10.4%), phenylalanine (9.9%), and glycine (9.0%). These collectively constituted 57.9% of the total number of amino acids. Amino acids encoded by T-rich codons (≥ 2 Ts in a triplet) were the most abundant and accounted for 42.7% of the total amino acid composition, whereas C-rich codons (≥ 2 Cs in a triplet) were the least used (they accounted for merely 5.0% of the total amino acid composition). As shown in Table 4, unequal usage of synonymous codons avoiding C at the third codon position was prominent in most cases; for instance, the relative frequency of using TTT for Phe was 9.2%, but the frequency of using TTC was only 1.4%.
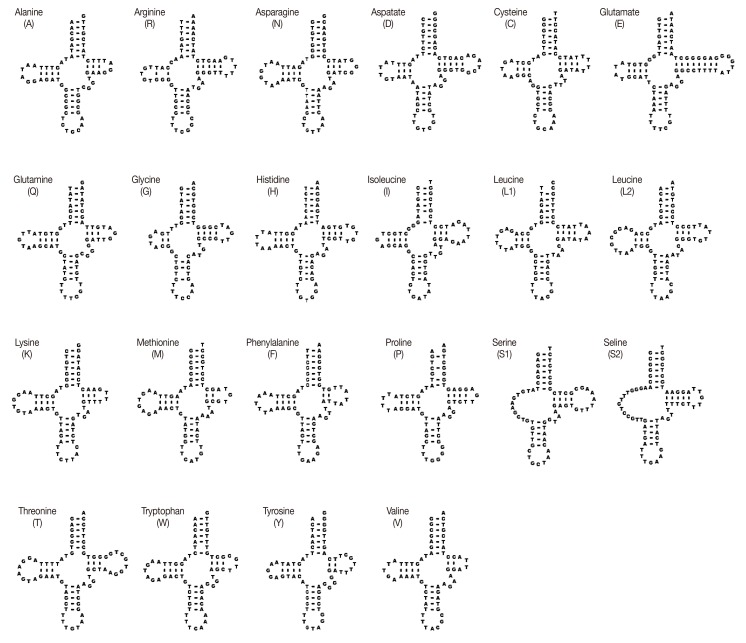
Transfer RNA genes, ribosomal RNA genes, and non-coding regions
The sizes of 22 tRNA genes identified in mt genomes of H. taichui ranged from 60 to 73 nucleotides (nt) in length. Of the 22 tRNA genes, 20 could be folded into the conventional cloverleaf structure, including a 7 bp amino-acyl stem, a 2-4 bp DHU arm with a 3-10 nt loop, a 5 bp anticodon stem with a loop of 7 nt, and a 2-8 bp of the TψC arm with a 3-10 nt loop. The 2 tRNAs specifying serine were exceptions (Fig. 2). The rrnL was located between tRNA-Thr and tRNA-Cys, and rrnS was located between tRNA-Cys and cox2. The rrnS and rrnL of H. taichui were 747 nt and 979 nt in length. Each ribosomal gene was assumed to directly abut neighboring genes. The A+T contents of the rrnL was 58.5%, respectively, and the A+T content of the rrnS was 55.9%. Just 1 long non-coding region (LNR) was identified, located between the tRNA-Gly and tRNA-Glu, and lacked any tandem repeats. The size was 1,710 bp, comprising 11.3% of the genome, and the A+T content was 58.3%.
Phylogenetic analysis
A concatenated alignment set of 3,380 homologous amino acid positions from conserved blocks was used. Phylogenetic relationships among the 15 flatworms using different analytical approaches (MP, ML, NJ, and BP methods) were the same in their topology (Fig. 3). Phylogenetic relationships among species were well resolved with very high nodal support throughout. In this tree, Schistosomatidae and (Fasciolidae+Paragonimidae+Opisthorchiidae+Heterophyidae) formed monophyletic groups. H. taichui was resolved as sister to Opisthorchiidae with a very high support in the phylogenetic analysis.
DISCUSSION
The mt genome arrangement of H. taichui was the same as that of Fasciola hepatica (NC_002546), Paragonimus westermani (NC_002354) and Opisthorchis spp., but distinct from the arrangement seen in some Schistosoma spp. [9,18]. All genes were transcribed in the same direction, as in other flatworms for which data are available [26]. H. taichui lacks atp8, a gene that is not seen in any flatworm species [18]. The majority of metazoan mtDNA sequences contain 2 non-coding regions of significant size difference, a long non-coding region (LNR) and a short non-coding region (SNR). In the case of H. taichui, a single long non-coding region was present. The nucleotide composition of the entire mtDNA of H. taichui was biased toward T and G, with T being the most common nucleotide and C the least favored, in accordance with mt genomes of other trematodes except for P. westermani (Table 3). Two genes, nad4L and nad4, overlapped by 40 nucleotides (Table 1), similar to the situation in other digeneans. The tRNA genes generally resemble those of other digeneans. A standard cloverleaf structure can be inferred for most tRNAs. Exceptions are tRNA-S in which the paired dihydrouridine (DHU) arm is missing in all parasitic flatworm species [18], although secondary structures including this arm are feasible for some species [15] including H. taichui. The rrnS was 747 nt in length, shorter than that of the homologs from other trematodes except for S. mansoni (744 nt) and P. westermani (744 nt, not registered on GenBank). The rrnS of S. mekongi was noted as being 709 nt (GenBank accession no. NC_002529), but it could be 39 nt longer if it directly abuts cox2 in that species. The rrnL of H. taichui, at 979 nt, was the shortest among trematodes recorded yet. Morphological data have traditionally been used for taxonomic studies on flatworms. Such data are now being supplemented by data from ultra-structural and biochemical studies [27,28] and, increasingly, from molecular sequences. Published phylogenies using nuclear ribosomal genes [19,29] indicate that the Heterophyidae is paraphyletic with respect to the Opisthorchiidae. Our mitochondrial sequences have yielded a tree consistent with this finding, with H. taichui seen as a sister to Clonorchis+Opisthorchis (family Opisthorchiidae). Sequences from additional heterophyid and opisthorchiid mt genomes will be required to confirm the findings from nuclear genes.
In conclusion, the present study reported the complete mtDNA sequence and genome organization of H. taichui for the first time. Its constituent genes were compared with homologs from other trematodes. Our phylogenetic analysis of concatenated protein-coding genes supports a sister group relationship between families Opisthorchiidae and Heterophyidae. These data will provide tools for the molecular diagnosis of haplorchiasis and for studies on the biology of the species.