INTRODUCTION
Traveler’s diarrhea (TD) shows symptoms of severe abdominal pain, urgent loose stool, vomiting, fever, and chills, and sometimes can lead to significant morbidity [1–3]. Currently, TD is the most common medical illness noted in international travelers: TD occurs in 15–50% of individuals traveling to high-risk regions including tropical or semitropical areas of America, southern Asia, Africa, and the Indian subcontinent [3,4]. The risk factors for developing TD include the lack of hygiene management in developing countries, rainy summer season, and the ingestion of contaminated food or drink [1]. High-risk foods for TD are usually associated with water contamination and include uncooked vegetables, unpeeled fresh fruit, uncooked meat, and seafood [1–3]. Outbreaks caused by waterborne protozoan parasites globally occurred between January 2004 and December 2010 and were caused by Cryptosporidium spp., Giardia lamblia, Toxoplasma gondii, Cyclospora cayetanensis, Entamoeba histolytica, and Blastocystis hominis [5]. While C. cayetanensis infection has been reported from numerous countries, it appears to be most commonly present in tropical and subtropical areas [6]. C. cayetanensis, C. parvum, and G. lamblia cause serious diarrhea for longer than 2 weeks after infection, and their poor response to standard antimicrobial treatment led to classify TD as antibiotic-resistant diarrhea [1]. Diagnosing which agent is reponsable for TD requires expensive laboratory detection methods and is time-consuming [3]. So far, only diagnostic methods based on simultaneous detection using real-time PCR analysis can save time and effort [7]. This study was designed to develop a simultaneous multiplex PCR detection method for identifying 3 waterborne protozoan parasites (e.g., C. parvum, G. lamblia, and C. cayetanensis) causing TD. In our previous study using conventional multiplex PCR, the amplified PCR products were distinguished by different band sizes, separated by gel electrophoresis [8]. However, this method is not quantitative and its primary advantage is the easiness of the experimental condition using primers and not probes [8]. The multiplex TaqMan-based real-time PCR method can distinguish each target using a different fluorescent dye, allowing to differentiate amplicons of similar size in a short time and with little effort [7]. In addition, the multiplex real-time PCR method was reportedly a sensitive and specific technique for detecting waterborne protozoan parasites [9]. With these features in mind, we aimed at developing a new multiplex real-time PCR detection method, which is applicable to stool specimens suspected of protozoa infection. In particular, the present study verified the efficiency of stool DNA extraction using BpT4 exogenously added because it has often a problem due to a poor efficiency of DNA extraction or incomplete removal of inhibitors when stool DNA was extracted [10,11].
We selected a target gene for each parasite, designed primers and probes in silico, and developed our detection method using primer-probe cocktail and parasite-spiked human stool samples. The present study using BpT4 as an internal control reports the successful development of a multiplex real-time PCR detection method with a primer-probe cockail able to detect each target DNA of 3 waterborne protozoan parasites.
MATERIALS AND METHODS
DNA preparation of parasites and virus
C. parvum oocysts and G. lamblia cysts were purchased from Waterborne™, Inc. (New Orleans, LA). C. cayetanenesis synthetic DNA containing the full 18S rRNA, ITS1, and ITS2 sequences was purchased from ATCC (ATCC® PRA-3000SD™, Manassas, Virginia, USA). BpT4 was purchased from CAROLINA™ (Carolina Biological Supply Company, Burlington, North Carolina, USA). For the preparation of genomic DNA for C. parvum oocysts, G. lamblia cysts, and BpT4, we used the DNeasy blood and tissue kit (Qiagen, Hilden, Germany). Briefly, parasites and BpT4 were subjected to repeated freezing and thawing cycles (8 cycles of 65°C for 1 min and liquid nitrogen for 30 sec) to break microorganism wall. DNAs extracted by the DNeasy kit were eluted with 20 μl buffer AE on the mini spin column and stored at −20°C. The purity and concentration of the extracted DNA were measured at 260/280 nm using a spectrophotometer (Nanodrop 2000, Thermo Scientific, Wilmington, Delaware, USA).
DNA extraction from human stool sample spiked by parasites and control virus
We prepared parasite-infected human stool samples by spiking oocysts of C. parvum and cysts of G. lamblia to non-infected human stool specimens. We confirmed that these human stool specimens were non-infected using parasite-targeted conventional nested PCR analysis and microscopy [8]. These non-infected human stool specimens were obtained from healthy screened participants for the preparation of the parasite and virus-spiked stool samples following approval by the Ethics Committee of the Inha University Hospital IRB (Research 15–026). The 1×106 oocysts and cysts of protozoan parasites (C. parvum and G. lamblia, respectively) were seeded into 1 g of uninfected human stool samples. Then, those samples were decimally diluted in PBS to obtain a dilution series containing 1×106-1×103 oocysts (cysts). In addition, the inoculation of BpT4 as an internal control for the stool DNA preparation was performed as follows. Purchased BpT4 was prepared in PBS at a concentration of 1×109 CFU/ml, and 10 μl (1×107 CFU) was seeded into each parasite-spiked stool sample. The DNA from the parasite and virus-spiked stool samples was extracted using the QIAamp® Stool Mini Kit (Qiagen) according to the manufacturer’s protocol. Briefly, stool samples were repeatedly frozen and thawed (8 cycles at 95°C for 1 min and liquid nitrogen for 30 sec) to break microorganism wall. The total genomic DNA was eluted using 20 μl of elution buffer (Qiagen). In the case of C. cayetanensis, we used purchased synthetic DNA because it was difficult to obtain the necessary amount of oocysts required for this experiment [8]. After 1×106 copies of synthetic DNA (ATCC) were serially diluted to 103 copies, 1 μl of the diluted DNA and 1 μl of the stool genomic DNA extracted from 200 mg of an uninfected stool sample were mixed and applied to the PCR reaction as a template.
Primer and probe design
PCR primers and probes for TaqMan real-time PCR analysis were designed to detect specifically targeted gene segments for each parasite and the virus (Table 1). The following target genes were selected: Cryptosporidium oocyst wall protein (COWP) of C. parvum, glutamate dehydrogenase (GDH) of G. lamblia, internal transcribed spacer 1 (ITS1) of C. cayetanensis, and gene product 21 (gp21) of T4 phage. The gene sequences of all primers and probes were newly designed in the present study using the Geneious version R8 software (Biomatters Ltd., Auckland, New Zealand). For the differential detection of each parasite, when 3 species of parasites are co-infected, 4 fluorescent dyes (FAM™, HEX™, Cy5™ and CAL Fluor Red® 610) were labelled to probes for Cp, Gl, Cc, and BpT4, respectively. The synthesis of primers and probes as well as the sequence analysis for amplicon identification were performed by Macrogen Inc. (Seoul, Korea) with the exception of CAL Fluor Red® 610-conjugated BpT4 probe performed by SFC Ltd. (Cheongju, Korea).
Development of protocols for multiplex real-time PCR analysis to simultaneously detect 3 protozoa species
Multiplex real-time PCR analysis was performed using a CFX96™ real-time PCR cycler (Bio-Rad, Philadelphia, Pennsylvania, USA). The amplification conditions were determined as follows: DNA templates (1–2 μl), 10 μl of 2×PCR premix (TOP-real™ qPCR 2×Premix, Enzynomics, Daejeon, Korea), 2 μl of the primer mixture (consisting of 4 pmol primers for each parasite), and 2 μl of the probe mixture (consisting of 4 pmol probes for each parasite) were mixed with HPLC water (Wako Pure Chemical, Osaka, Japan) in a total volume of 20 μl. The absence of non-specific binding of the prepared primers and probes was confirmed by real-time PCR analysis of the genomic DNA of the uninfected human stool sample using a primer-probe set for each parasite as well as a primer-probe cocktail for the 3 protozoa and BpT4. The multiplex real-time PCR conditions consisted of 15 min at 95°C for pre-incubation, followed by 40 cycles of denaturing at 95°C for 10 sec with annealing and extension at 60°C for 1 min. Bio-rad CFX manager software (Bio-rad) was utilized to generate the standard plots for each DNA target. To correct the difference of Ct value due to the potential effect of the copy number of one of the target genes on the result of quantitative real-time PCR, we adopted a conversion formula between the number of oocysts (cysts) and copies number of each gene. LOD of each parasite in the stool sample was represented by the Ct value based on copy number (not oocyst or cyst number) and calculated by the following formula: (Estimated DNA copy number in test sample amount for one-time PCR analysis=N×C×D1×D2; with N =number of oocysts (cysts) in the stool sample, C=known copy number of target gene, D1=dilution factor of stool weight, D2=dilution factor of DNA volume: used volume for reaction/total eluted volume. For example, COWP gene of C. parvum and GDH gene of G. lamblia exist each as a single copy in these parasites’ genomes [12,13]. As the copy number of the ITS1 gene is unknown in C. cayetanensis, we estimated it to be 100 copies based on the evolutional phylogenetic tree similarity with the rDNA gene of other protozoa (Toxoplasma gondii and Eimeria tenella previously reported having 100 copies for the ITS1 gene) [14]. The LOD in the present study was represented by the Ct value in each parasite.
RESULTS
Target specificity of the designed primers and probes for the development of the multiplex real-time PCR detection method
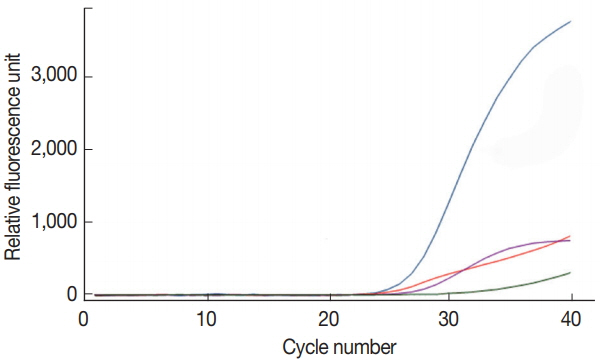
To confirm the target specificity of the primer pairs newly designed in the present study, a single real-time PCR reaction was performed for each DNA of C. parvum, G. lamblia, and C. cayetanensis, and the amplified PCR products were analyzed by DNA sequencing. The amplified PCR products showed 100% similarity to AB089292 (C. parvum), KJ499992 (G. lamblia), and AF302546 (C. cayetanensis) reported in the GenBank database (http://www.ncbi.nlm.nih.gov) (data not shown). A primer-probe cocktail did neither show any non-specific amplification on the negative control stool DNA, nor on other protozoan parasites (i.e., Entamoeba histolytica, Toxoplasma gondii, and Babesia microti) used for checking cross-reactivity (data not shown). In Fig. 1, the primer-probe cocktail detected specifically and individually each target DNA (COWP, GDH, ITS1, and gp21 gene from C. parvum, G. lamblia, C. cayetanensis, and BpT4, respectively) contained in the protozoa DNA mixture of C. parvum, G. lamblia, C. cayetanensis, and BpT4. The protozoa DNA mixture included the genomic DNA of 105 oocyst (cyst) of C. parvum and G. lamblia, 105 copies of the synthetic DNA of C. cayetanensis, and 107 CFU of BpT4; the primer-probe cocktail included 4 pairs of forward and reverse primers and 4 probes for the 3 protozoa species and BpT4. The fluorescent dyes FAM™, HEX™, Cy5™, and CAL Fluor Red® 610 probed well the target DNA of C. parvum, G. lamblia, C. cayetanensis, and BpT4, respectively, and were detected by the color blue, green, purple, and red, respectively (Fig. 1). Furthermore, there is no cross reaction between primers and protozoa DNA, and the negative control without template does not show any amplification of DNA (Fig. 1). This result shows that our protocol for multiplex real-time PCR analysis using newly prepared primer-probe cocktail is an appropriate method to detect simultaneously 3 protozoa species.
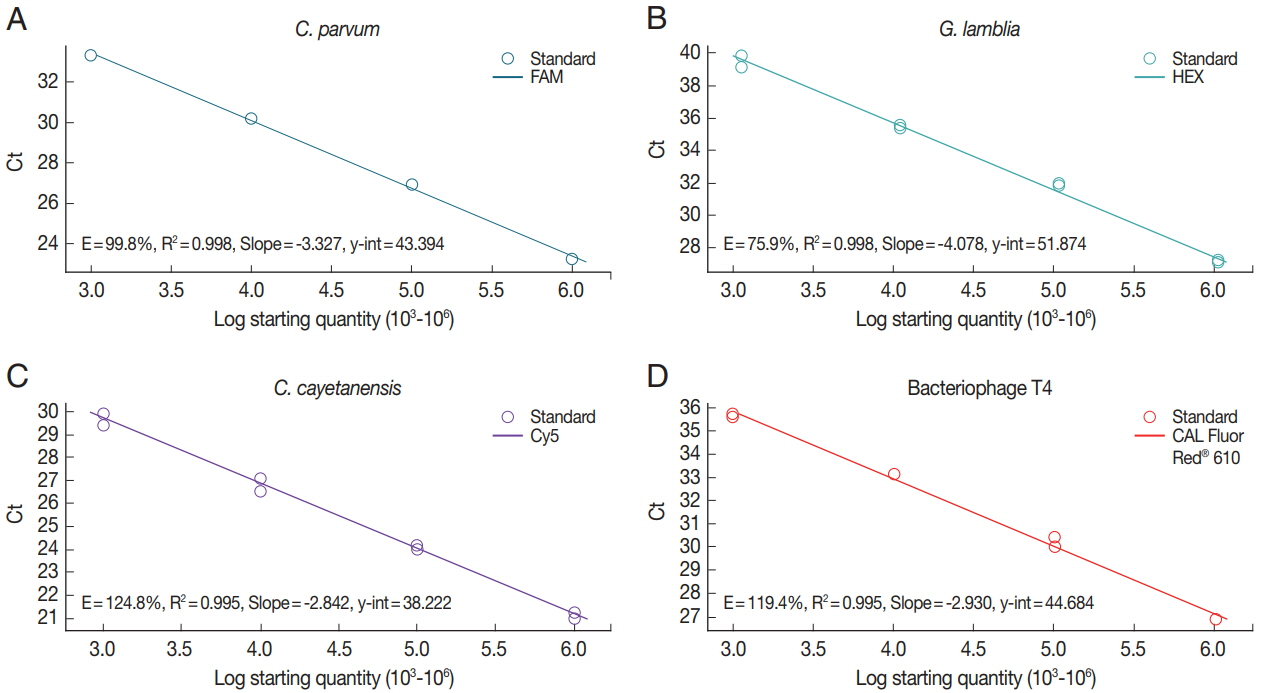
Standard curve of the concentration of parasite spiked in human stool samples
To investigate the sensitivity of real-time PCR analysis of the parasite concentration in the stool samples, we determined each standard curve representing the Ct values by parasite concentrations (Fig. 2). Genomic DNAs isolated from C. parvum (Fig. 2A)- and G. lamblia (Fig. 2B)-spiked stool samples (106 oocyst [cyst]/stool [g]), mix of genomic DNA of uninfected control stool and synthetic DNA of C. cayetanensis (106 copy/stool [g]) (Fig. 2C), or 106 CFU of BpT4 (Fig. 2D) were serially diluted 10 times and evaluated using our protocol and a primer-probe cocktail. R-square values of the regression line and efficiency of the PCR reaction were analyzed using Bio-rad CFX manager software (Bio-rad). Real-time PCR results (Ct value) by parasite concentrations are represented by a reverse regression line dependent on the parasite concentration. We found that the R-square values for the multiplex real-time PCR method using a primer-probe cocktail range between 99.5% and 99.8%. This result shows that real-time PCR analysis using a primer-probe cocktail can detect each parasite concentration depending on the genomic DNA of each parasite-spiked stool sample and BpT4-spiked stool sample. In addition, this standard curve can be used to calculate the parasite concentration of an unknown stool sample.
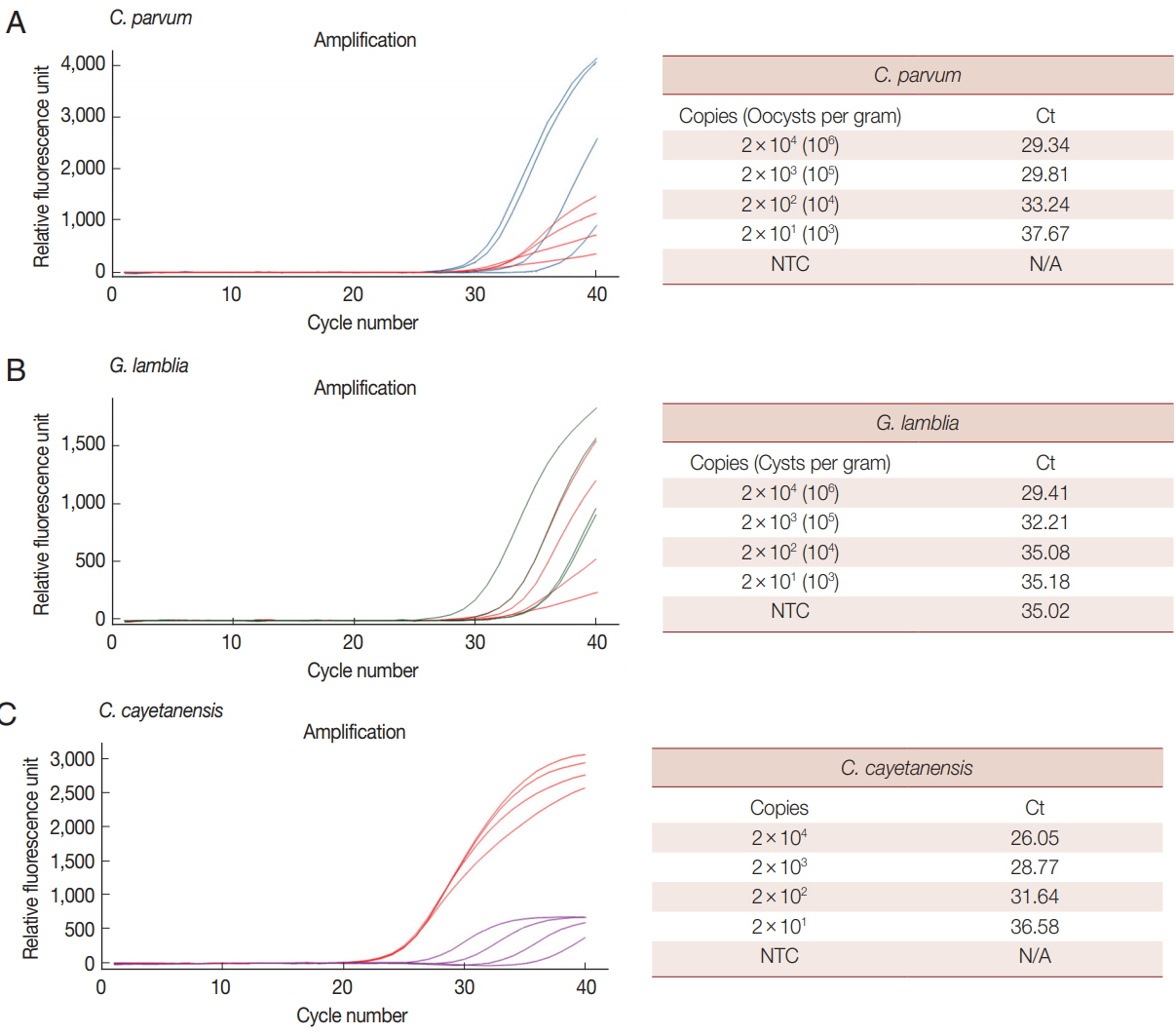
Limit of detection of each parasite in the parasite-spiked human stool samples
We analyzed the LOD of our multiplex real-time PCR detection method to evaluate the LOD in clinical specimens (Fig. 3). C. parvum and G. lamblia were prepared at concentrations ranging from 1×106 to 1×103 oocysts (cysts). Each parasite concentration was spiked to human stool samples along with the BpT4 internal control (107 CFU), and genomic DNA was extracted from the stool (Fig. 3A, B). In the case of C. cayetanensis, 1 μl of each synthetic DNA diluted previously from 1×106 to 1×103 copies was mixed with parasite-free stool DNA and BpT4-spiked stool DNA (Fig. 3C). These template DNAs were reacted with a primer-probe cocktail, and the results are shown by Ct values (Fig. 3). Because PCR amplification of target DNA is proportional to the copy number of target DNA, we converted the oocyst (cyst) number in parasite-spiked stool sample (g) into the copy number of target DNA in the sample amount to be tested (20 mg of stool sample) as described in the “Materials and Methods” section.
When 1×106 to 1×103 of oocysts (cyst) of C. parvum and G. lamblia were spiked in human stool samples (g), their copy numbers in the sample amount to be tested were converted into 2×104 to 2×101, respectively (Fig. 3). The LOD for each concentration of parasite is expressed by the Ct value (Fig. 3). In this test, 107 CFU of BpT4 were spiked in all stool samples as an internal control. The LOD of C. cayetanensis is also presented as a Ct value for the copy number (2×104 to 2×101) in each sample amount to be tested (Fig. 3). Our results show that the Ct values for C. parvum, G. lamblia and C. cayetanensis were 29.34, 29.41, and 26.05, respectively, at the parasite concentration of 2×104 copies, and they were 37.67, 35.18, and 36.58, respectively, at the parasite concentration of 2×101 copies. From the above results, the LOD of C. parvum and C. cayetanensis in stool samples was determined to be 2×101 copies. The negative control without template (NTC) does not show any DNA amplification during the 40 cycles of amplification demonstrating the target specificity of primers and probes developed in the present study (Fig. 3). However, the LOD of G. lamblia was determined to be 2×103 copies because the NTC shows non-specific binding (Fig. 3). While the COWP gene for C. parvum and the ITS1 gene for C. cayetanensis are very sensitive target genes, the GDH gene for G. lamblia may be a less sensitive target gene to diagnose parasite infection in real-time PCR analysis using a primer-probe cocktail. However, the advantage of the GDH gene for targeting G. lamblia remains that there was no cross reaction to C. parvum and C. cayetanensis. Accordingly, the combination of the 3 target genes is adequate for the multiplex real-time PCR detection method.
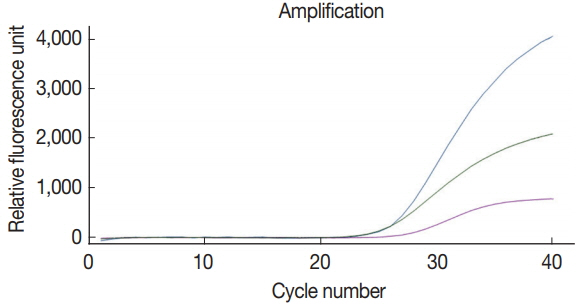
Simultaneous detection of 3 species of parasites using multiplex real-time PCR analysis with a primer-probe cocktail
The multiplex real-time PCR method was tested for the simultaneous detection of 3 species of parasites using a primerprobe cocktail. We prepared a DNA mixture of C. parvum, G. lamblia, and C. cayetanensis composed of genomic DNAs of 107 oocysts (cysts)-spiked stool samples for C. parvum and G. lamblia as well as the mixed DNA of 105 copies of C. cayetanensis and negative control stool DNA. The multiplex real-time PCR detection method developed in the present study was effective to amplify simultaneously the 3 species of parasites (Fig. 4). Probes of C. parvum, G. lamblia, and C. cayetanensis conjugated with reporter dyes (FAM™, HEX™, and Cy5™, respectively) specifically detected each parasite signal without interference or non-specific signal. Thus, our protocol for multiplex real-time PCR is an applicable method to diagnose simultaneously 3 protozoan parasites, C. parvum, G. lamblia, and C. cayetanensis, in human stool samples.
DISCUSSION
We developed a novel detection method using TaqMan® probes for simultaneously identifying target DNA of the waterborne protozoan parasites C. parvum, G. lamblia, and C. cayetanensis. Methods to detect waterborne protozoa include microscopic examination, immunology-based methods, fluorescence, and in situ hybridization [15]. Currently, PCR is the most commonly used technique and is more sensitive than microscopic examination [16]. PCR-based strategies that can diagnose simultaneously enteric pathogens including waterborne protozoan parasites and foodborne bacterial pathogens have been developed for multiplex PCR, real-time PCR, nucleic acid sequence-based amplification, and loop-mediated isothermal amplification [9,17,18]. Multiplex PCR is a type of PCR method, which uses more than one pair of primers in order to identify simultaneously several target sequences [8,19]. The method saves time and effort by concurrently amplifying multiple target sequences in one reaction and lowers the cost of the reaction while increasing the throughput [8,20].
The more recently developed multiplex real-time PCR is more sensitive and specific for detecting water borne protozoan parasites in fecal samples than the conventional multiplex PCR method [9]. Real-time PCR method can reduce labor and time-consuming steps for detecting PCR products through gel electrophoresis analysis [7]. Additionally, real-time PCR presents the advantage to not require to take into consideration the DNA size of target genes as fluorescence-tagged probes are used, and also to minimize the contamination due to the closed tube system for testing [21,22]. In this respect, real-time PCR allows theoretically to detect simultaneously several target genes with different fluorescence-tagged specific probes. However, practical difficulties include fluorescent signal interference, simultaneously detected targets limitation, and difficulty to detect more than 2 different targets [21]. Because developing a multiplex real-time PCR method is difficult, our protocol enabling multiplex real-time PCR detection for C. parvum, G. lamblia, and C. cayetanensis is helpful for diagnosis. This protocol can test the type and amount of protozoa infection in clinical samples as it amplified individually a DNA fragment of each target gene and quantified each amount in human stool samples.
Stool specimens are complex mixtures frequently containing amplification inhibitors and commensal bacteria resulting in false-negative diagnoses [23]. In our method, we used T4 phage as an internal control to monitoring stool DNA amplification [24]. Bacteriophage T4 is inexpensive to produce, nonpathogenic, and quantifiable, making it an ideal internal control [24]. Our real-time PCR results with T4 phage as an internal control confirm its value as internal control for detecting the protozoa target DNA in our tests. The design of primers and probes has played a key role in the success of the method. The sequences of specific primers and probes were compared among several target genes of the 3 protozoan parasites via in silico analysis using the Geneious software (version R8) to ensure specific amplification and avoid cross-reactivity with DNA fragments from enteric bacteria. We selected COWP as target gene for C. parvum, GDH for G. lamblia, and ITS1 for C. cayetanenesis because these genes are frequently used for classifying parasite species and confirming parasite isolates [25–27]. Our goal was to appropriately mix them after we determined target sequences to play the roles of specific primers and probes. As expected, the developed primer-probe cocktail was able to detect the specific target genes simultaneously in human stool samples in which the 3 species of parasites were co-spiked.
Primers and probes for the target genes in C. parvum and C. cayetanensis display a good sensitivity with LOD values of 2×101 copy (corresponding to 1×103 oocysts g−1 of feces for C. parvum). Previously, the detectable oocysts number from symptomatic cryptosporiosis patients has been reported to be 1×105–1×107 oocysts g−1 of feces for C. parvum [28]. With the sequence variability of C. cayetanensis ITS1 sequence, primers and probe determined in the present study have a superior target specificity and sensitivity with an LOD value of 2×101 copy [27,29]. Even though primers and probes targeted to the GDH gene in the stool sample was less sensitive as shown by the LOD of G. lamblia, the GDH gene is highly specific and sensitive to G. lamblia with a consistency of 95.7% between microscopy and PCR diagnosis [30]. Most importantly, the primerprobe cocktail developed in the present study specifically detected each parasite signal without any interference or nonspecific signal in the human stool sample.
Taken together, our method can detect simultaneously 3 different diarrhea-causing protozoan parasites in stool samples. Although further studies are required to evaluate the sensitivity and specificity using clinical samples for developing diagnostic assays, our multiplex real-time PCR detection method is the first attempted TaqMan-based real-time PCR method for the simultaneous detection of C. parvum, G. lamblia, and C. cayetanensis using T4 phage as an internal control.