Trichomonas vaginalis infection is the most prevalent non-viral sexually transmitted infection worldwide. The World Health Organization (WHO) estimated that 248 million people were infected with T. vaginalis in 2005, and by 2008 this number had increased by 11.8% to 276.4 million [1]. According to recent studies, women infected with T. vaginalis are at high risk of vaginitis, endometritis, atypical pelvic inflammatory disease, and adverse pregnancy outcomes [2,3]. In men T. vaginalis increases the risk of infertility and prostate cancer [4]. Moreover, T. vaginalis infection has been linked with increased risk and transmission of HIV [5–7]. Given the clinical significance of T. vaginalis infection, accurate diagnosis is required.
Various methods have been used to diagnose trichomoniasis, such as wet mount and culture. Wet mount examination is straightforward and rapid, but more than 103/ml live protozoa are needed, and culture requires inocula of 300–500 trichomonads/ml and a specialized medium, as well as taking 2–5 days [8]. Because of these limitations, a highly sensitive and specific PCR method has been used in recent years [9,10]. In fact, multiplex PCR assays have been used extensively to diagnose sexually transmitted infections (STIs), because they allow for faster detection, and reduce labor and reagent costs [11,12]. The Seeplex_STD6 auto-capillary electrophoresis (ACE) detection assay employs 6 pairs of dual priming oligonucleotide (DPOTM) primers specifically targeting unique genes of Chlamydia trachomatis, Neisseria gonorrhoeae, Mycoplasma genitalium, Ureaplasma urealyticum, Mycoplasma hominis, and Trichomonas vaginalis [12].
The present study aimed to compare the sensitivity for detecting T. vaginalis infections of the Seeplex® ACE Detection Kit (Seegene, Seoul, Korea) with that of a PCR assay, HY-PCR, developed in our laboratory.
A total of 4 urine samples were used, 2 from BPH patients and 2 from prostate cancer patients. The 4 Korean patients were being treated in the Department of Urology, Hanyang University Hospital. The study was approved by the Institutional Review Board of Hanyang University, and written informed consent was obtained from the patients and controls (IRB No. HYUH 2013-04-028-006).
First-voided urine (VB1) specimens were used in the PCR assays [10]. The VB1 samples (30–40 ml) from the 4 patients were provided in sterile 50 ml screw-cap plastic tubes, immediately frozen at −20°C, and transported on ice to the laboratory where they were thawed and centrifuged for 10 min at 1,500 g. The supernatants were aspirated, and the sediments were transferred to tubes containing 3 ml PBS.
DNA was prepared by 2 methods; following the instructions with the QIAamp® DNA Mini Kit (Qiagen, Valencia, California, USA), and heat-treating with a chelating resin (Chelex) [13]. The manufacturer’s instructions for the QIAamp® DNA Mini Kit were as follows; 200 μl washed urine sample was mixed with 20 μl Protease K, and 200 μl Buffer AL was added to 200 μl of sample and incubated at 56°C for 10 min. 200 μl ethanol was added and the mixture was applied to a QIAamp® Mini spin column (in a 2 ml collection tube) and centrifuged at 6,000 g for 1 min and the tube containing the filtrate was discarded. 500 μl Buffer AW1 was added to the Mini spin column and centrifuged at 6,000 g for 1 min, and the tube containing the filtrate again discarded. The Mini spin column was placed in a clean 2 ml collection tube, and 500 μl Buffer AW2 was added and the column was centrifuged at full speed (20,000 g) for 3 min, and the tube discarded. 200 μl Buffer AE was added to the Mini spin column and centrifuged at 6,000 g for 1 min. Five μl of Buffer AE containing the eluted DNA was used as DNA template. Thus, the process for isolating DNA with the QIAamp kit involves several steps.
PCR reaction mixtures for the Seeplex® ACE Detection Kit assays contained 5×STD6 ACE PM (primer) 4 μl, 8-MOP solution 2.5 μl, 2×multiplex master mix 10 μl, STD IC 1 μl, template 3 μl. The DNA was initial denatured for 15 min at 94°C, followed by 40 cycles of 30 sec denaturation at 94°C, 1 min 30 sec annealing at 63°C, and 1 min 30 sec extension at 72°C.
DNA for the HY-PCR was prepared by heat-treating with a chelating resin (Chelex) [13]; 1 ml of suspended urine precipitate was centrifuged at 10,000 g for 5 min, resuspended in 200 μl of a 5% suspension of chelating resin (Chelex® 100; Sigma, St. Louis, Missouri, USA) in 0.01 M Tris buffer (pH 8.0), and incubated at 56°C for 30 min. The preparation was mixed gently, boiled for 10 min, and centrifuged at 12,000 g for 5 min in a microcentrifuge, and 5 μl of supernatant was used directly as the template for PCR. The primers were based on the T. vaginalis-specific repetitive DNA sequence in clone TV-E650-1 [14].
The primers used were: 5′ gagttagggtctaatgtttgatgtg 3′ and 5′ agaatgtgatagcgaaatggg 3′. The PCR reaction mixtures contained 1 μl each of the primers at 10 pmol/μl each, 2 μl dNTPs (2.5 mM each), 0.1 μl Taq polymerase (5 U/μl), 5 μl DNA, 2 μl 10×PCR buffer, 5.2 μl 5 M betaine, and 3.7 μl distilled water. The DNA was denatured for 5 min at 94°C, followed by 40 cycles of 10 sec denaturation at 98°C, 30 sec annealing at 55°C/52°C, and 30 sec extension at 72°C.
To avoid product carryover, the PCR reactions were set up in an area physically separated from all activities involving amplified target sequences, thermocycling, and running of gels. To assess the sensitivity of the PCR, a suspension of T. vaginalis was counted with a hemocytometer and numbers of trophozoites were adjusted with PBS to 1, 10, and 100 per PCR mixture. To confirm whether or not the 318 bp band obtained by PCR originated from T. vaginalis, nested PCR was undertaken using primers: 5′ATCCCC-AACAATGAACGAAG 3′ and 5′AATGTGATAGCGAA-ATGGGA 3′; this confirmed the 181 bp band.
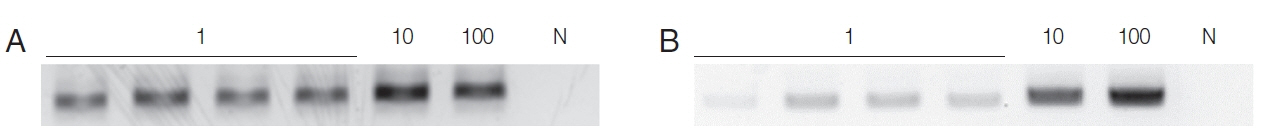
When the HY-PCR assay was performed with DNA extracted with Chelex® 100 and with the QIAamp® Mini Kit, the HYPCR assay with DNA extracted by either method gave a positive result with samples containing 1 trophozoite although the latter DNA gave a weaker band than the former (Fig. 1). This result suggests that Chelex® 100 is more effective at extracting DNA than the QIAamp® Mini Kit.
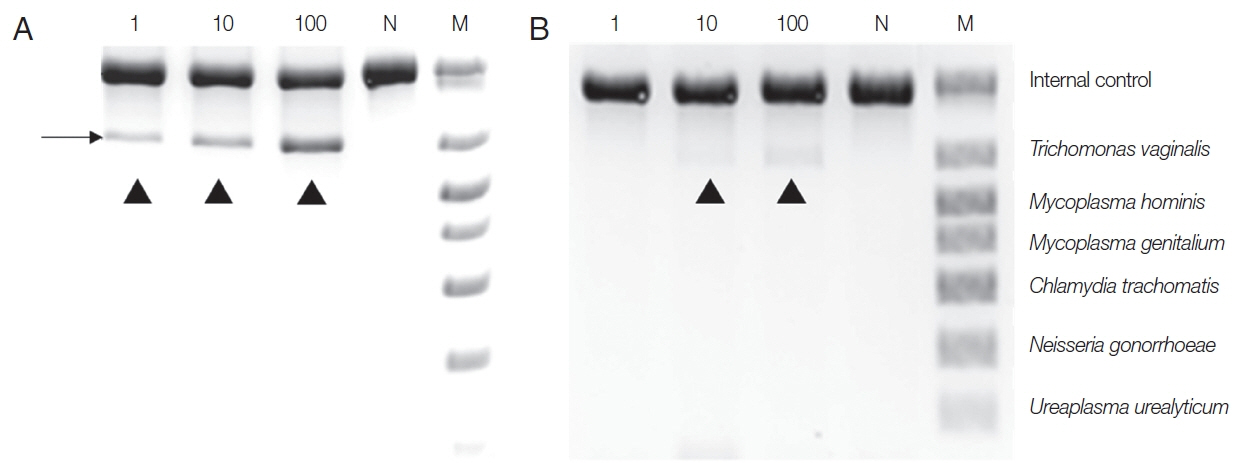
To make the conditions similar to those in samples from patients, T. vaginalis trophozoites (1, 10, 100) were added to the urine of a normal man and incubated overnight, and DNA was extracted with Chelex® 100, or a QIAamp DNA Kit, and PCR was performed with a Seeplex® ACE Detection Kit. DNA from the Chelex® 100 gave a positive finding from samples containing 1 trophozoite. In contrast, the DNA exacted with the QIAamp® Mini kit gave only the faintest positive band from 10 trophozoite samples, and 100 T. vaginalis samples yielded dimly visible band (Fig. 2). Therefore, the sensitivity of the Seeplex® ACE Detection Kit PCR seemed to vary depending on how DNA was extracted, and Chelex® 100 was again superior to the QIAamp® Mini Kit for extracting DNA.
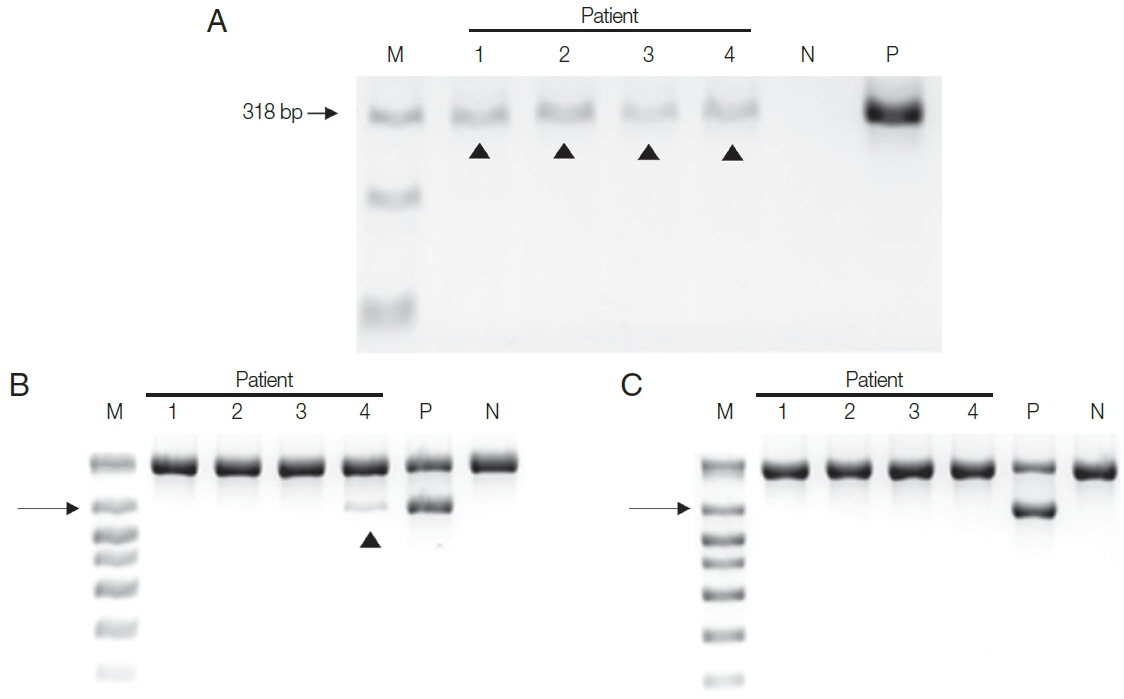
Next, DNA extracted from the four patient samples was used in the two PCR systems. HY-PCR gave positive findings with all 4 urine samples when using the Chelex®100 (Fig. 3A). By contrast, Seeplex® PCR gave positive findings with only one of the four samples using the Chelex®100 (and none with the DNA made with the QIAamp® Mini Kit) (Fig. 3B, C). In other words, only negative results were obtained in Seeplex® PCR with DNA made with the QiAamp Kit recommended by the Seeplex PCR Kit. Evidently HY-PCR using DNA extracted with Chelex is highly sensitive not only when using cultured trichomonads but also when using patient samples.
However, Seo et al. [15] reported the lower limit of PCR system developed in Kyungpook National University using Tvk3/7 and BTUB9/2 primer set was 1 organism per reaction. When the prevalence of Tv in 201 men attending a Primary Care Urology Clinic was tested with this PCR system, eight men (4%, 8/201) yielded positive bands. In contrast, all eight gave negative results with the Seeplex® Kit PCR with DNA made with the QiAamp Kit. These findings are consistent with our results.
Lee et al. [12] have discussed the possible limitations of multiplex PCR, which include PCR drift due to stochastic fluctuations in the interactions between PCR reagents and competitive inhibition by PCR selection. Despite this limitation, the Seeplex® Kit PCR assay is widely used in clinical practice because it can detect six different pathogens causing sexual transmitted infections at the same time and is cost effective. This multiplex PCR kit was found to be highly sensitive for detecting T. vaginalis because a distinct positive band was observed from a single trophozoites when DNA was isolated by the Chelex method. All our results show that the DNA extraction method influences the sensitivity of the Seeplex PCR system. Interestingly, the QIAamp® Mini Kit did not perform better than Chelex® 100 despite its higher cost. The QIAamp® Kit was also one of the more labor-intensive methods tested in the current study, as it included three different incubation temperatures and eight centrifugation steps [16]. Considering that the Seeplex® ACE Detection Kit is used in most of Korean hospitals, the DNA extraction method recommended by the kit should be modified to increase its sensitivity for detecting T. vaginalis.
A limitation of this study is that it involved a small number of patient samples. The examination of more samples is needed to ensure confidence in its findings.
In conclusion, considering its clinical importance, there is an urgent need to develop a highly sensitive diagnostic method for T. vaginalis. The present findings should help in this direction.