INTRODUCTION
The larval stage of the tapeworm Taenia pisiformis (Cestoda: Taeniidae) is the causative agent of cysticercosis in rabbits, and is globally distributed [1,2]. T. pisiformis cysticercosis usually parasitizes the liver capsule, gastric omentum majus, and mesentery of rabbits, while adult T. pisiformis establish and mature in the small intestine of dogs and foxes. China is the world's largest producer of rabbits [3], and T. pisiformis has become one of the most common parasites to severely affect rabbit breeding. It mainly causes autologous poisoning and emaciation, but can also weaken resistance to other diseases; it may even cause death [4]. However, insufficient studies on genetic variation of T. pisiformis in China have been carried out to date. Due to faster mutation rates of mitochondrial DNA (mtDNA) sequences than nuclear genes [5] and the absence of host selection pressures [6], mtDNA sequences are considered to be more suitable to discriminate between closely related organisms [7].
Mitochondrial genes have successfully been used to study genetic variation, and enable a focus on the genetic origin, scope, and genotype of organisms [8]. The structure and function of cytochrome b (cytb) have been verified in mtDNA sequences of cestodes and maintain a moderate evolutionary speed. Thus, cytb has been used to study the population structure and genetic differentiation of several tapeworm species [9,10]. In this study, we determined the genetic variation of T. pisiformis based on partial cytb gene sequences from Sichuan Province, China.
MATERIALS AND METHODS
Sample collection
A total of 53 isolates were collected from routine autopsies in 8 geographical regions of Sichuan Province, China. The regions included Ya'an (7 isolates, YA1-YA7), Chengdu (7 isolates, CD1-CD7), Panzhihua (6 isolates, PZ1-PZ6), Leshan (7 isolates, LS1-LS7), Guangyuan (7 isolates, GY1-GY7), Luzhou (7 isolates, LZ1-LZ7), Guang'an (6 isolates, GA1-GA6), and Aba (6 isolates, AB1-AB6) (Fig. 1). The maintenance and care of the rabbits used in this study was in strict accordance with good animal practice regulations.
DNA extraction and PCR conditions
Approximately 0.5 g genomic DNA was extracted from cysticerci using the phenol-chloroform extraction as described by Sambrook et al. [11]. The DNA was resuspended in 50 µl Tris-EDTA (TE) buffer and stored at -20℃. To amplify the cytb gene, PCR primers (forward: 5'-ATGGTTAGTTTATTACGTCGGA-3'; and reverse: 5'-TAAGAACTCTAAACACTTGACATAC-3') were designed by the program primer 5.0 using the mitochondrial genome sequence of T. pisiformis, which is available at the National Center for Biotechnical Information (NCBI) database (GenBank no. NC013844). PCR reactions were carried out in a 50 µl reaction mixture containing 25 µl of PCR mixture (Tiangen, Beijing, China), 2 µl of each primer (forward and reverse), 1 µl of template DNA (approximately 100 ng), and 20 µl sterile water. The amplification conditions for cytb consisted of an initial denaturation step at 94℃ for 5 min, followed by 30 cycles of denaturation at 94℃ for 55 sec, annealing at 54℃ for 55 sec, elongation at 72℃ for 50 sec, and a final extension step at 72℃ for 10 min. PCR products (50 µl) were separated by electrophoresis on a 1.0% agarose gel and stained with ethidium bromide. Amplicons were cloned into a pMD19-t vector (TaKaRa, Dalian, China) according to the manufacturer's instructions. Purified PCR products and positive clones were sequenced 3 times in-house using an ABI PRISM™ 377XL DNA Sequencer (ABI, Foster City, USA) with universal forward and reverse primers, respectively.
Sequence analysis
The sequences of the cytb gene were confirmed by a comparison with the published mitochondrial genome sequence of T. pisiformis. Multiple alignments were performed in ClustalX 1.83. Aligned sequences (excluding primer sequences) were transformed to FASTA and MEGA files. The number of haplotypes, calculation of haplotype diversity (Hd), nucleotide diversity (π), gene flow (Nm), and neutrality tests (including Tajima's D, and Fu and Li's test) were performed using DnaSP 4.10.9 [12]. To obtain the conserved and variable sites, global FST value and AMOVA were analyzed using Arlequin v3.11 [13]. Network 4.0 software [14] was used to analyze the MJ-network of haplotypes. The phylogenies were reconstructed using the neighbor-joining (NJ) method in MEGA 4.0 [15]. Parameters for tree construction included the Kimura-2-parameter index and 1,000 bootstrap resampling.
RESULTS
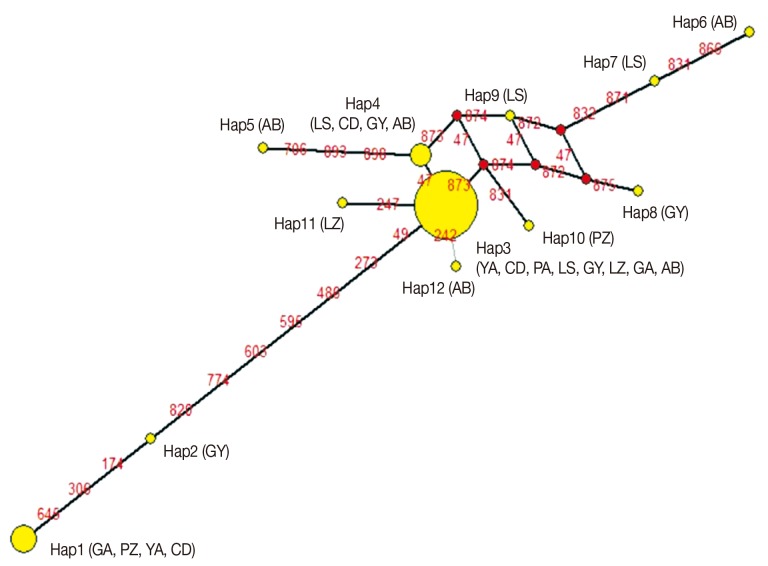
The partial sequence size of the cytb gene was 922 bp, which occupied 86.3% (922/1,068) of the whole length. The sequences of 53 isolates were submitted to GenBank under the accession numbers JN870153-JN870178 and JX535256-JX535282. The average base composition of cytb was 44.9% (T), 26.3% (A), 19.9% (G), and 8.9% (C), with AT-richness in the sequences. No insertions, deletions, or stop codons were detected. Twenty-four variable sites, including 17 parsimony informative sites and 7 singleton sites, were found in cytb. These included 14 transitions (12 A-G and 2 T-C), and 10 transversions (3 A-T, 2 A-C, 1 G-C, and 4 T-G). Twelve haplotypes were found in 53 isolates, giving an Hd value of 0.578 and a π value of 0.00371. As observed in this study, Hap3 was the most common haplotype (found in 35 isolates, 66.0%), followed by Hap1 (YA2, GA1, GA2, CD1, and CD2) and Hap4 (AB2, LS2, CD3, and GY3). Nine haplotypes were found only in a single isolate, including GY4 (Hap2), AB3 (Hap5), AB1 (Hap6), LS3 (Hap7), GY2 (Hap8), LS1 (Hap9), PZ3 (Hap10), LZ7 (Hap11), and AB6 (Hap12).
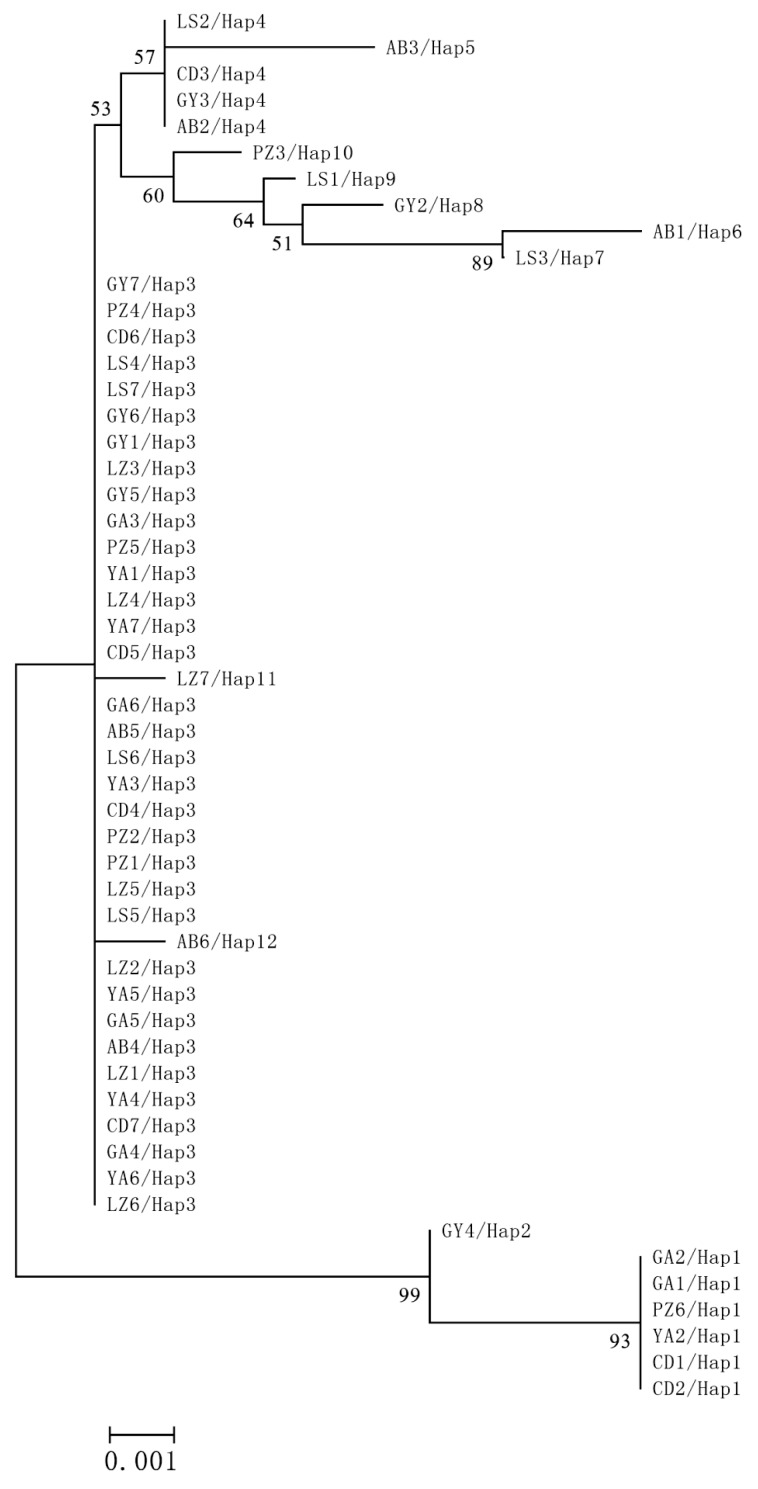
The AMOVA showed that 98.4% genetic variation was derived from intra-region, and 1.6% from inter-region. The global Nm and FST of genetic differentiation across 53 samples were 1.46 and 0.0157, respectively. Analysis of the haplotype MJ-network showed that Hap3 was the most prominent haplotype, and the other haplotypes centered on it (Fig. 2). No geographical clustering was observed from the NJ tree analysis (Fig. 3). However, haplotype clustering was significant in the phylogenetic trees. The first cluster was Hap1 and Hap2, and the second was Hap3 to Hap12. The neutrality test showed that Tajima's D=-1.13902 (P>0.1), Fu and Li's D*=-0.52626 (P>0.1), Fu and Li's F*=-0.88276 (P>0.1) were not significantly different.
DISCUSSION
In our study, the AT-richness of cytb exceeded 70%, which was similar to observations by Jia et al. [16] and Liu et al. [17]. They suggested an AT-bias in the mitochondrial genomes of T. taeniaeformis, T. multiceps, T. hydatigena, and T. pisiformis. The π value is an important index to measure the level of genetic diversity. In most animals, a π value of >0.01 is considered to indicate a comparatively large variation [18]. In this study, π value was lower than 0.01, suggesting that there was low genetic variation of 53 isolates from 8 regions. According to the different allele frequency, the genetic differentiation index, FST, is often used to evaluate the proportion of genetic diversity [19]. Nm>1 is considered enough to resist the function of genetic drift, and prevent the occurrence of genetic differentiation [20]. Nm value (1.46) and FST (0.01573) of global isolates demonstrated low levels of genetic diversity. The 53 geographcial isolates may belong to a larger population in Sichuan Province. There was no evidence demonstrating a geographical clustering from the haplotype MJ-network and phylogeny tree. Phylogenetic analyses revealed that there was no correlation between phylogeny and geographical distribution. In our study, the values of Tajima's D, Fu and Li's D, and Fu and Li's F scores proved that the evolution of T. pisiformis followed a neutral mode.
Barbosa et al. [21] used the wild rabbit (Oryctolagus cuniculus) and a parasitic tapeworm (T. pisiformis) as an example to study phylogeographic triangulation, and argued that the phylogeography of a parasite that needs 2 hosts to complete its life cycle should reflect population history traits of both hosts. Many macroparasites do not actively disperse their populations but rely on the movement of their intermediate or final hosts [22,23]. Following the increasingly frequent trade of canines and rabbits over long distances, the exchange of genetic material appears more likely, which may cause the low genetic variation of T. pisiformis in Sichuan Province, China.
In conclusion, our results showed that low genetic variations were present in 53 isolates of T. pisiformis from Sichuan, China, which provided genetic evidence for future development of prevention and control measures. However, the relationship between T. pisiformis pathogenicity, drug resistance, and genetic structure requires further research to clarify the role of each.