INTRODUCTION
Cryptosporidium is a unicellular microscopic parasite that infects a wide range of vertebrate hosts including humans [1]. Typically, this pathogen is enteric, shed in feces, and commonly transmitted through drinking water causing diarrhea [2]. Laboratory diagnosis of cryptosporidiosis is commonly established through microscopic examinations of fecal smears stained with acid fast dye for the presence of oocysts [3]. Certain oocysts surface antigens, captured by synthesized monoclonal antibodies, have been used as targets of detection with immunoassays-based methods. Unlike these methods, PCR is a more sensitive tool not only for parasite detection but for characterization as well [3].
Several PCR-based molecular assays have been developed for detection of Cryptosporidium infection in human feces since decades. Most of these assays have been executed without internal standards that give assurance of the PCR-negative results. PCR-negative results may be true-negative or false-negative due to its amplification failure [4,5]. Being an enzymatic reaction, PCR may be constrained by organic or inorganic substances in clinical samples. Fecal samples, in particular, can contain many of these substances [4-6]. Other substances that could inhibit PCR may be accidentally brought to reactions during the sample processing or nucleic acid extraction steps [4]. PCR can be partially or completely inhibited by one or another of these substances. Partial inhibition decreases PCR sensitivity while complete inhibition causes false-negative results. Both results represent major problems in clinical laboratories and have significant negative impacts on patient management as well as infection control.
PCR inhibition could be avoided or minimized by an appropriate selection of sample processing procedure, a nucleic acid extraction protocol, a stable polymerase enzyme or use of specific PCR additives [4-6]. Besides these useful measures, PCR inhibition has to be monitored by a suitable internal amplification control (IAC) to avoid false-negative results, especially in clinical laboratories as previously recommended [7].
IAC is a non-target nucleic acid sequence that is simultaneously co-amplified with the primary target sequence in the same PCR tube [7]. Internal control could be designed to monitor the sample preparatory step (s) alone [8] or PCR amplification step alone [9-11], or both steps [12,13]. IAC may be designed to be competitive or non-competitive. The competitive IAC shares the target DNA sequence in the reaction constituents including the primers. As a result, 2 amplicons of various molecular weights are produced for each positive sample [7,8]. On the contrary, the non-competitive IAC has a nucleic acid sequence which is distinct from the target DNA sequence and is amplified by another primer pair different from that is designed to amplify the primary target sequence [14].
In an earlier study, an extraction protocol based on QIAamp® DNA Stool Mini Kit has been developed for protozoan DNA extraction, including Cryptosporidium, directly from the diarrheic stool specimens. The DNA yield has been sufficiently purified and proved compatible with diagnostic PCR [15]. Thus, no PCR inhibition has been described in all control samples subjected to amplification. Together with the subsequent IAC-free Cryptosporidium PCR assay, it has been applied on a panel of random fecal samples and as a result, many samples have been diagnosed as Cryptosporidium-negatives. Questions about the level of confidence for these previously prescribed findings, have served as a motivation for construction of an appropriate internal standard for the Cryptosporidium PCR assay in the current study. To achieve this goal, we (i) presented an in-house constructed IAC, (ii) incorporated it in the assay with an estimated optimal concentration, (iii) evaluated it using the same panel of control as well as random fecal samples that have been previously examined with the IAC-free PCR assay, and (iv) compared the results before and after the IAC integration into the assay.
MATERIALS AND METHODS
Types of samples
The PCR assay was tested on different samples including clinical samples and purified oocysts. As a source of genomic DNA and to estimate the analytical performance of the PCR assay, a purified preparation of 8×105 Cryptosporidium parvum oocysts with PBS in volume of 1 ml was purchased from Moredun Animal Health (Scotland, UK). To estimate the diagnostic performance of the PCR assay, 25 Cryptosporidium-positive and 45 Cryptosporidium-negative control fecal samples were prepared in this study using a combined gold standard test comprising modified Ziehl Neelsen (mZN) microscopy, RIDA® Quick Cryptosporidium (R-Biopharm, Darmstadt, Germany) and 18S rRNA nested PCR [16]. In addition, 180 fecal samples were randomly collected for evaluation of the PCR assay. All feces were collected from samples submitted for diagnosis to various hospitals in Al-Taif, Saudi Arabia.
DNA extraction and PCR amplification
DNA extractions from feces, oocysts suspensions, and oocysts-spiked feces were carried out using the QIAamp® Stool Mini Kit (Qiagen, Valencia, California, USA) following the amended kit’s protocol [15]. Plasmid DNA was purified from the transformed cells with QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany). Amplification reactions of the reference nested PCR [16], inverse PCR [17], colony PCR [18], and the PCR, under study [19] were carried out following the previously published protocols. Amplifications were done using TechneTM TC-4000 thermal cycler. The GoTaq® Hot Start Polymerase (Promega, Madison, Wisconsin, USA), and other reagents were used in reactions with final concentrations closely similar to the published protocol. PCR products were analyzed on 1-2% of agarose gel electrophoresis.
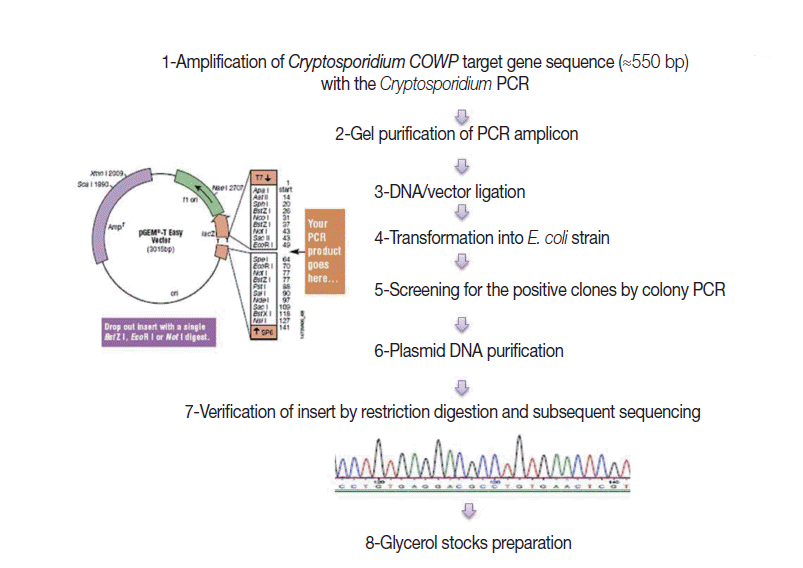
Construction of external amplification control (EAC) plasmid
Fig. 1 shows steps adopted for construction of the EAC. Briefly, a genomic C. parvum DNA sample, extracted from the purified oocyst suspension, was subjected to amplification by the target PCR assay. Gel-purified PCR products (≈550 bp), with the YORBIO Gel/PCR DNA Purification Kit (Yorkshire Bioscience, York, UK) were cloned into the pGEM®-T-Easy cloning vector (Promega). After transformation into TOP10 Escherichia coli competent cells (Invitrogen, Carlsbad, California, USA), 10 white colonies after overnight incubation were selected and screened for the correct plasmid by colony PCR as previously described [20]. Plasmid DNA was purified and its concentration was determined as previously described [20]. Insert verification was carried out by EcoRI (New England Biolabs, Hitchin, UK) restriction digestion (RD) and bidirectional automated sequencing (Eurofins MWG, Ebersberg, Germany). For subsequent PCR use, 1 ng DNA Stock was prepared and the copy number of the COWP gene sequence was calculated following a previously described equation [21].
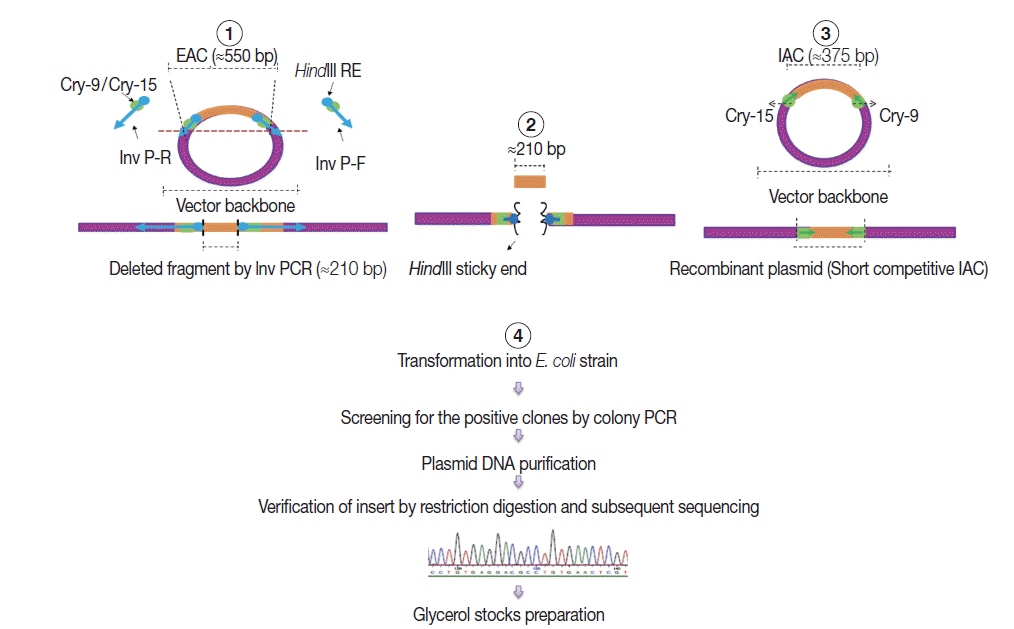
Construction of internal amplification control (IAC) plasmid
As shown in Fig. 2, a short competitive IAC (375 bp) was prepared by deleting a DNA fragment between the Cry-9 and Cry-15 primers using a certain type of PCR called inverse PCR (Inv PCR) as described by Ochman et al. [17] as follows: The retrieved EAC plasmid sequence was found free from unique restriction enzyme sequence. Therefore, 2 inverse primers were manually designed with additional HindIII restriction enzyme recognition sequence plus 2 additional nucleotides were incorporated at the 5΄ terminus of each primer. The Inv PCR was set up and as a result, ≈210 bp was deleted using the following primers; Inv P-F (ATA AGC TTA TTG ATA TGG TCT GCC CAC C΄) and Inv P-R (TTA AGC TTA AAA CCA GAA GGA CAA ACG G). The gel purified amplicons (≈3.4 kb) were subsequently, subjected to restriction digestion (RD), with DpnI (New England Biolabs), and HindIII (Roche, Sussex, UK). The Inv PCR-truncated plasmid was re-ligated on itself and transformed into TOP10 E. coli competent cells. DNA stock was prepared and stored at -20˚C for subsequent PCRs, similar to EAC Plasmid.
Optimal concentration of the IAC
The optimal concentration of IAC was determined by a 2-Step experiment as follows: Firstly, 1 ng of EAC and IAC was serially diluted down to concentrations of 0.04 fg and 0.1 fg per μl, subsequently. PCR was run for EAC dilutions, as a sole amplification target and the lowest dilution giving amplicon on gel was defined. Secondly, the defined EAC concentration was included with each IAC dilution in 1 PCR tube as a duplex PCR. The optimum IAC concentration was defined as the lowest dilution that was consistently detectable with EAC on gel. All subsequent amplifications were run as duplex PCR with the optimum IAC concentration determined.
Analytical sensitivity of the PCR assay
Aliquots of Cryptosporidium-free feces, 200 μl of each, containing ≈1,700, 1,500, 1,000, 500, 100, 50, and 10 of the purified C. parvum oocysts were prepared. DNA extracts were subjected to PCR amplification in the presence of IAC as duplex reactions and the lower detection limit (LDL) for the PCR assay was estimated.
Diagnostic performance of the PCR assay
Based on the results of the combined gold standard test, control fecal samples, of various oocysts densities were prepared as follows: Samples diagnosed as Cryptosporidium-positive (n=15) by microscopy as well as the reference PCR were considered of high oocysts load. Samples diagnosed as Cryptosporidium-negative by microscopy but positive with immunoassay and the reference PCR test (n=7) were considered of moderate oocysts load. Samples with low oocysts density (n=3) was considered for those were positive by PCR only. Finally, Cryptosporidium-negative samples (n=45) were negative by the 3 tests. DNA extracts of these samples (n=70) were subjected to amplification with the PCR assay.
Validation of the PCR assay on random fecal samples
Randomly-collected fecal samples (n=180) were screened for Cryptosporidium by mZN microscopy, RIDA® Quick Cryptosporidium and the PCR assay. Samples with discordant results were re-tested with the 18S rRNA reference PCR. Absence of IAC amplicon on gel for 1 sample was considered false-negative and was re-tested.
RESULTS
Constructed plasmids
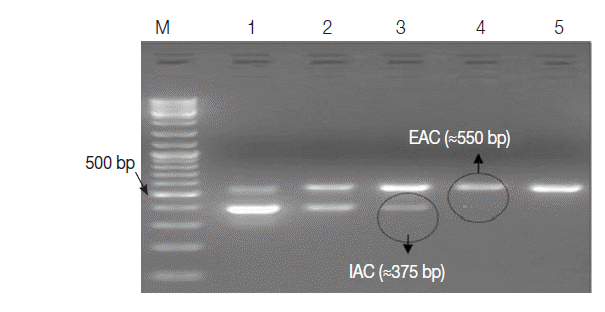
Two recombinant plasmids were successfully developed and preserved in E. coli bacterial strain as glycerol stocks for future usage. Some important features for each plasmid were shown in Table 1. The EAC with the lowest concentration of 0.4 fg per μl (≈100 copies) was sufficient to be clearly detected by the assay as an external positive control for the PCR. The optimum concentration of IAC was estimated to be about 1 fg/reaction (Fig. 3).
Analytical sensitivity of the PCR assay
The Cryptosporidium DNA was successfully extracted and amplified, in parallel with the IAC target, from all fecal samples seeded with C. parvum oocysts down to a concentration of about 100 oocysts per stool extract (200 mg), corresponding to 2 oocysts per PCR.
Diagnostic sensitivity of the PCR assay
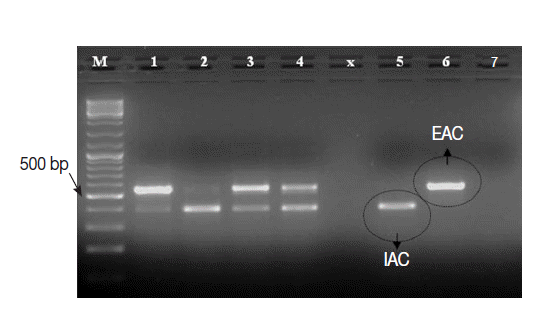
The Cryptosporidium native target DNA was successfully amplified, side by side with the IAC, in all Cryptosporidium-positive control samples (n=25), except 1 sample. This sample with false negative result showed IAC amplicon on gel and was 1 of the 3 samples with low oocysts density. Interestingly, the intensity of IAC (≈375 bp) and the native (≈550 bp) PCR products alternately appeared abnormally faint on gel in 2-3 samples (Fig. 4). None of the Cryptosporidium-negative control samples (n=45) showed amplification of the native COWP gene by the PCR assay. However, the IAC amplification products were successfully detected on gel for all samples. On view of these findings, the PCR assay was found to exhibit sensitivity, specificity, negative predictive value and positive predictive value of 96%, 100%, 98%, and 100%, respectively. Equally important, none of control samples DNA extracts showed detectable inhibition for the PCR amplifications.
Validation of the PCR assay on random clinical samples
Out of the 180 samples, Cryptosporidium was detected in 21 (11.6%), 18 (10%), and 17 (9.4%) samples by the PCR, RIDA® Quick, and microscopy, respectively. Samples with discordant results (n=4) were Cryptosporidium-positive by the 18S rRNA reference PCR. IAC PCR product was exhibited on gel for all samples.
DISCUSSION
In the current study, an internal control was successfully designed to improve the clinical utility of the previously studied Cryptosporidium diagnostic PCR assay. As has been mentioned earlier, many strategies can be adopted for IAC construction. Selection among these strategies is usually based on many factors including the desired purpose, PCR type and length of the PCR product [22].
In our study, construction of IAC was carried out by deleting a number of nucleotides internal to the primers attaching sites of the cloned PCR product. According to previous studies, modification of the target DNA sequence may be accomplished through deleting or inserting a number of nucleotide sequences internal to the PCR primers [14]. This could be done straightforward if a unique restriction enzyme sequence, present in the cloned DNA target but not in the cloning vector backbone was present. In the present study, this unique restriction enzyme sequence was absent. Thus, insertion of a HindIII restriction enzyme sequence inside the PCR target sequence was mandatory. The inv PCR, as strategy for IAC construction, has been previously adopted but with approach that is different from that followed in this study [23]. In the current study, the inv PCR was used to insert the HindIII restriction enzyme's sequence inside the cloned PCR target sequence and to delete a certain number of nucleotides. The enzyme's sequence was inserted as an overhanging sequence at the 5΄ terminus of each inverse primer. Two additional nucleotide bases were added at the 5΄, distal to the enzyme's recognition sequence, to provide stability of DNA-enzyme complex and facilitate efficient cutting of sites located close to the ends of the linear DNA as previously explained [24]. Prior to ligation, the Inv PCR amplicon was firstly treated with DpnI that selectively cuts only the methylated DNA removing any remaining of the template plasmid DNA [25] and secondly with the HindIII restriction endonuclease to facilitate ligation by producing sticky ends. Storage of the recombinant plasmids in competent bacterial cells offers the continuous availability of high quality IAC DNA with controlled stability, size and high copy number. The size of IAC was selected to be close to that of the native target DNA to reduce the primers preference towards 1 target over the other as previously reported [26].
Another important finding of this study was the adequate feasibility of the constructed IAC. When spiked with the target DNA sample, 2 PCR products of various molecular weights were developed and easily analyzed on gel till a certain concentration described as the optimum concentration. In this study, the PCR assay was re-tested against DNA samples of various purities. On its integration into the Cryptosporidium diagnostic PCR while doing re-testing for Cryptosporidium-positive control fecal samples, both PCR targets were successfully amplified but with minimal competition noticed on gel for few number of samples. The reduced intensities of the PCR products, alternated between the IAC and the native target, on gel were apparent for 2-3 Cryptosporidium-positive control fecal samples with various oocysts load. In spite of the observed competition between IAC and the primary PCR target towards the shared reaction kinetics, including the primers, the analytical as well as the diagnostic sensitivity of the assay was not altered by integration of IAC [15].
In view of the literature, the mean number of oocysts shed by a Cryptosporidium-infected person has been reported to be ≈3.3×106 oocysts per ml of stool during symptomatic infection and ≈3×105 oocysts per gram of stool for asymptomatic cases [27]. Thus, any competition that may occur between the IAC and the native PCR target, when applied as duplex PCR on diarrheal feces, will be in favor of the later. Taking all together, the Cryptosporidium PCR with the inhibition control proved sensitive, properly standardized assay and were useful for detection of Cryptosporidium infection in humans.
Equally important finding, all the Cryptosporidium-negative samples obtained in the previous study [28] proved to be true negatives. Only the IAC amplification bands were successfully noticed on gel for all samples. This adds more confidence to the capability of the DNA extraction protocol that has been previously developed to extract and purify the protozoan DNA directly from human feces [15].
A variety of exogenous (non-competitive) IAC has been adopted in few previous real-time PCR assays. In these assays, a control signal, instead of amplicons on gel, is always produced when there is no target sequence in the test sample. In 2013, Yang et al. [9] have used a fragment of a coding region from Jembrana disease virus (a cattle disease) in 1 assay. Hadfield et al. [10] have used a commercial IAC in another assay. The phocin herpes virus 1 (PhHV-1) has been adopted as IAC for real-time multiplex PCR assays detecting Giardia and Entamoeba histolytica in parallel with Cryptosporidium in human feces [12,13,29]. In spite of the reported high sensitivities (1-10 oocysts/PCR), in many cases, real-time assays are often hampered by the high cost of the thermal cyclers, extra primers and PCR reagents. Moreover, the virus that has been spiked into feces prior to extraction is unlikely to be a suitable extraction control for Cryptosporidium protozoan DNA because the oocysts are undoubtedly much more resistant to extraction than this lipid enveloped herpes virus. Persson et al. [30] have used the broad-range bacterial 16S ribosomal DNA-based primers to amplify bacterial DNA found in fecally derived crude DNA samples as an IAC for non-bacterial diagnostic PCR assays. However, due to the massively high copy number of the 16S ribosomal DNA (IAC) relative to the non-bacterial targets sought in these enteric PCR assay, the PCR reaction kinetics would be heavily biased towards IAC amplification. Hence, amplification of the IAC sequence could not rule out, with any confidence, the presence of inhibitory substances present in the fecally derived DNA samples.
In conclusion, an IAC control, of competitive trait, was constructed for Cryptosporidium diagnostic PCR assay to monitor the amplification. This IAC was found to be very helpful in ruling out PCR inhibition for all the fecally-derived crude DNA samples without alteration of the performance of the PCR assay. All results that have been given by the IAC-free Cryptosporidium diagnostic PCR assay were identical to that obtained in the current study with IAC/target PCR assay. Therefore, Integration of IAC into the Cryptosporidium diagnostic PCR assay added more assurance to the previous results.