INTRODUCTION
Malaria is a major tropical disease that is responsible for 0.4–0.5 million deaths worldwide [1], with the vast majority of severe and fatal cases being confined to pregnant women and children of less than 5 year of age [2,3]. To reduce the burden of malaria, effective vaccines against the respective parasites, especially for Plasmodium falciparum, are indispensable tools. Vaccines against the pathogenic blood stage of the malaria parasites would be useful not only in speeding up parasite clearance, but also in limiting the spread of anti-malarial drug resistance, which is now highly prevalent in Southeast Asia and many parts of the world [4]. One of the major obstacles to vaccine development is the antigenic diversity of the genes in natural parasite populations, as demonstrated by whole genome sequencing [5], which allows the parasites to escape host immune responses. The proteins of blood stage malaria parasites, such as merozoite surface proteins, have been identified as promising targets of immune responses, but they are reported to be highly polymorphic [6]. Because a high degree of polymorphism occurs in field isolates, the polymorphism of the antigen-coding genes must be extensively studied before the development of an effective vaccine can be accomplished.
One of the leading candidate antigens for the development of a blood (erythrocyte) stage vaccine against P. falciparum is the apical membrane antigen 1 (AMA-1). The AMA-1 protein is initially localized at the apical end of the merozoite that binds to the erythrocyte [7]. In addition, AMA-1 has been shown to be expressed in the sporozoite, which is the stage that participates in hepatocyte invasion [8]. The full-length protein is comprised of 622 amino acid residues, has a molecular weight of 82 kDa, and can be divided into the 2 regions of a 550-amino acid extracellular region at the N-terminal and a transmembrane region at the C-terminal [9]. The extracellular domain (ectodomain) is further divided into 3 domains (domains I, II, and III) and contains 16 Cys residues forming 8 intra-molecular disulfide bonds responsible for maintaining the 3D structure of proteins [10]. The location of the disulfide bridges are highly conserved in all Plasmodium species. Upon egression of the merozoite from an infected erythrocyte, the N-terminal region (prodomain) of the AMA-1 protein is cleaved into a 66 kDa fragment, which is translocated to the neck of rhoptry through interaction with rhoptry neck protein 2 (RON2) [11]. Subsequently, during the merozoite invasion of erythrocytes, secondary proteolytic cleavage occurs, resulting in the 48 kDa and 44 kDa fragments [12]. Disruption of this proteolytic processing with antibodies against AMA-1 has been shown to inhibit erythrocyte invasion [13]. Furthermore, examination of the binding activity of P. falciparum AMA-1-derived peptides revealed that the ectodomain of AMA-1 possesses the erythrocyte binding activity and is a putative target of protective immune responses [14]. The C-terminal portion of the protein is membrane-bound and carried into the invaded erythrocyte. Although the cytoplasmic tail of AMA-1 is not required for the correct trafficking and surface translocation, it has been shown to be essential for AMA-1 functions [15].
Currently, AMA-1 recombinant vaccines based on the P. falciparum 3D7 and FVO strains are being tested in Phase 1 and 2 clinical studies, and the vaccines have shown promising results [16–20]. Nevertheless, one study using antibodies raised against recombinant AMA-1 from P. falciparum strain 3D7 (the genome reference strain, [21]) revealed that the anti-AMA-1 antibodies were potent inhibitors of 3D7 homologous strains but were less effective against strains with heterologous AMA-1 haplotypes [22]. This suggested that sequence variation within the AMA-1 gene allows the parasites to escape the host immune responses, thereby posing a significant challenge for vaccine design. Accordingly, an understanding of the natural variation within the AMA-1 gene sequence in the parasite populations will have an important practical implication for the design of future AMA-1-based vaccines against P. falciparum. AMA-1 is encoded by a single-copy, intron-less gene that spans a 1,869 base pair (bp) region on chromosome 11 (PF11_0344) [21]. As a vaccine candidate, the genetic diversity of the AMA-1 gene has been a subject of intensive research. Detailed molecular studies on the polymorphism of the P. falciparum AMA-1 gene have been conducted in various endemic areas worldwide, including the Middle East and South Asia [23–28], Oceania [29–31], Africa [31–39], and South America [40–41]. Whereas the majority of the AMA-1 sequences spanned the regions of domain I or the ectodomain (domains I-III) of the gene, there are currently only limited datasets in which the full-length sequences of the AMA-1 gene are reported [31,40,42]. In addition, there is only limited AMA-1 sequence data from P. falciparum populations in Southeast Asia, including the Thailand-Myanmar border and none from the Thailand-Cambodia and Thailand-Laos borders, where the multidrug resistant parasites are often detected and the vaccines will be needed most [43,44]. Rather, within this region, AMA-1 sequence data is mainly derived from small local parasite populations in Western Thailand [31,42,45] and near the China-Myanmar border [46,47]. Furthermore, none of these studies have combined the data to generate a global view of the AMA-1 diversity in P. falciparum populations in Thailand.
To address this deficiency, this study aimed to determine the sequence diversity of the full-length sequences of AMA-1 in P. falciparum collected from 5 geographical locations near Thailand’s national borders. Comparative genetic analysis of the AMA-1 gene between P. falciparum isolates in Thailand and those of global geographical regions were performed. The outcome of this study will provide an insight into the nature and evolution of AMA-1 polymorphisms, which is a key feature for vaccine development.
MATERIALS AND METHODS
Origins of the P. falciparum malaria parasite isolates used in this study
Natural parasite isolates of P. falciparum used in the present study were originally obtained from 68 patients in 5 malaria endemic localities in Thailand, including Mae Hong Son, Kanchaburi, Ranong, Trat and Ubon Ratchatani (Supplementary Fig. 1). The parasite collections were conducted between 2001 and 2010. The blood stage malaria parasites were subjected to in vitro cultivation and maintained at the Department of Biology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand. The species of the malaria parasites were confirmed by microscopic examination of thin blood films and by genotyping with genetic markers, including microsatellites [48] and the merozoite surface protein-1 (MSP-1) and merozoite surface protein-3 (MSP-3) genes [49,50].
Merozoites of P. falciparum were expanded in the laboratory using a in vitro culture protocol. Once the levels of parasitemia reached 5–10%, the parasites were harvested and used for genomic DNA extraction using a phenol/chloroform extraction method [48–50]. In brief, the parasitized erythrocytes were lysed in 0.05% (w/v) saponin in phosphate buffered saline pH 7.4 and then incubated in lysis buffer overnight. The cell lysates were treated sequentially with (i) saturated phenol, (ii) 25: 24: 1 (v/v/v) phenol: chloroform: isoamyl alcohol and (iii) chloroform prior to addition of 3 M sodium acetate and absolute ethanol for precipitation of the genomic DNA. The dried DNA pellets were dissolved in TE (10 mM Tris.HCl/1 mM EDTA, pH 8.0) buffer and stored at -20°C until use.
PCR and DNA sequencing
The PCR reaction for amplification of the P. falciparum AMA-1 gene was prepared in a 50-μl volume containing 5–10 ng genomic DNA, 0.5 μmol of each of the For-AMA1 and Rev-AMA1 primers (Supplementary Table S1), 0.5 mM MgCl2 and 1 unit of Faststart Taq DNA polymerase (Roche, Mannheim, Germany) in 1× Taq buffer. Thermal cycling was performed at 95°C for 10 min, followed by 40 cycles of 95°C for 1 min, 60°C for 1 min and 72°C for 2 min, and then a final 72°C for 10 min. The PCR products were analyzed by agarose gel electrophoresis. The PCR amplicons generated from the genomic DNA of 68 parasite isolates that produced a single PCR amplicon were then submitted to commercial DNA sequencing at Bioneer (Daejeon, Korea) using the primers shown in Supplementary Table S1. DNA sequencing was performed bidirectionally using BigDye v3.1 and analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, California, USA). The obtained DNA sequences were aligned and contigs were assembled using Sequencher V. 4.5 (Gene Codes). The quality of DNA sequences and all SNPs were assessed by visual inspection to ensure that there were no gaps in the alignment.
Population genetic analysis
Full-length (−1.8 kb) sequences of AMA-1 (n=134 alleles) from P. falciparum populations in Thailand were obtained from the nucleotide databases at the NCBI [31,32,40,42] (Supplementary Table S2). The AMA-1 sequences by Polley et al. [45] were not included in the population genetic analysis because the only 1.3-kb sequences were available. Each unique sequence of P. falciparum AMA-1 was treated as a unique haplotype, and the haplotype files were created using the DnaSP 5.0 software [51]. Average number of pairwise nucleotide differences (K), nucleotide diversity (π), number of haplotypes (H) and haplotype diversity (Hd) were calculated using MEGA software [52]. The π estimated average number of substitutions between any 2 sequences, assuming that the sample is random. The distribution of the genetic diversity across the AMA-1 gene was also plotted using a sliding window with a window length of 100 bp and moving the window in steps of 3 bp in the DnaSP 5.0 software to identify the region(s) of AMA-1 that had accumulated polymorphisms. The π values were calculated in each window, and the values of π from each window were then plotted against the nucleotide position.
The mean number of non-synonymous substitutions per non-synonymous site (dN) and synonymous mutations per synonymous site (dS) within each isolate were estimated using Nei and Gojobori’s method [53], with the Jukes and Cantor correction [54], as implemented in the MEGA software. The significance of any differences between dN and dS were tested with the Z-test of selection (P <0.05), where a dN/dS ratio of greater than 1 at the 95% confidence interval was taken as evidence of positive diversifying selection. In addition, 3 population genetic tests of neutrality (Tajima’s D, and Fu and Li’s D* and F* tests) were used for detecting the signature of positive selection, that is whether polymorphism in AMA-1 sequences occurred at higher or lower frequencies than expected under a neutral model [55,56]. Sliding window plots, with a window length of 100 bases and a step size of 3 bp, were also generated if there were specific regions with a significant departure from neutrality (P <0.05).
Analysis of linkage disequilibrium was performed between nucleotide sites at which the frequency of the minor allele was >0.1 using an R2 index with Fisher’s exact test of significance. The minimum number of recombination events in the history of samples was estimated according to the method of Hudson and Kaplan [57]. All calculations for the neutrality tests, linkage disequilibrium and population recombination parameters were performed in the DnaSP 5.0 software.
Population differentiation analysis
The P. falciparum AMA-1 sequences from natural isolates in Asia, Oceania, Africa and South America were downloaded from the NCBI database (Supplementary Table S2). Nucleotide sequences corresponding to domain I from positions 445–906, corresponding to amino acid residues 149–302, were included in the population differentiation analysis. Arlequin suite version 3.5 software was used to calculate Wright’s fixation index (Fst) to evaluate the differences in the distribution patterns (ratios) of AMA-1 haplotypes between different geographic populations of P. falciparum [58]. Statistical significance was accepted at a P-value of <0.05.
Construction of the AMA-1 gene tree
The analysis involved 47 nucleotide sequences (unique haplotypes) of P. falciparum AMA-1 in Thailand and one AMA-1 sequence from the chimpanzee malaria parasite Plasmodium reichenowi (NCBI accession no. AJ252087; [59]) that served as an out group. There were a total of 1,815 positions in the final dataset (from nt positions 25–1,838). The evolutionary history was inferred using the maximum likelihood (ML) method based on the general time reversible model. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining (NJ) and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach, and then selecting the topology with a superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 46.4463% sites). The evolutionary history was inferred based on the general time reversible with the Hasegawa-Kishino-Yano with gamma distribution shape parameter (HKY+G) model. Codon positions included were 1st+2nd+3rd+Noncoding. The reliability of the ML tree was accessed by the bootstrap method with 1,000 pseudo-replicates. Evolutionary analyses were conducted in MEGA 7.
RESULTS
Nucleotide sequence analysis of AMA-1
The PCR amplifications of AMA-1 from DNA templates of 68 P. falciparum isolates all yielded a single PCR amplicon with an estimated size of 1,800 bp. and were then sequenced. DNA sequences from 3 parasite isolates from Mae Hong Son province (MH07, MH09, and MH28) had ambiguous nucleotide sequences, likely due to the presence of multiple infections, and so were excluded from the analysis. Thus, the 1,848-bp nucleotide sequence fragment of the AMA-1 gene, corresponding to nucleotide positions 10–1,857 of P. falciparum 3D7, from 65 P. falciparum isolates in Thailand were analyzed.
Multiple sequence alignment of AMA-1 identified 60 single nucleotide polymorphisms (SNPs). Of these, 58 and 2 SNPs resulted in non-synonymous and synonymous mutations, respectively. 4 sites (nucleotide positions 586, 590, 598, and 922) were trimorphic loci, while the remaining 56 sites were dimorphic loci. The 60 SNPs were distributed in 51 polymorphic codons (Supplementary Table S3). Forty-three codons encoded dimorphic amino acids, 6 codons with 2 SNPs encoded trimorphic amino acids, while the other codon with 2 SNPs encoded 4 types of amino acids. In addition, there was one codon with 3 SNPs encoding 6 different amino acids. Although the majority of these SNPs have been reported in earlier studies [31,42,45], an SNP at nucleotide position 730 (GAT, TAT) is reported here for the first time.
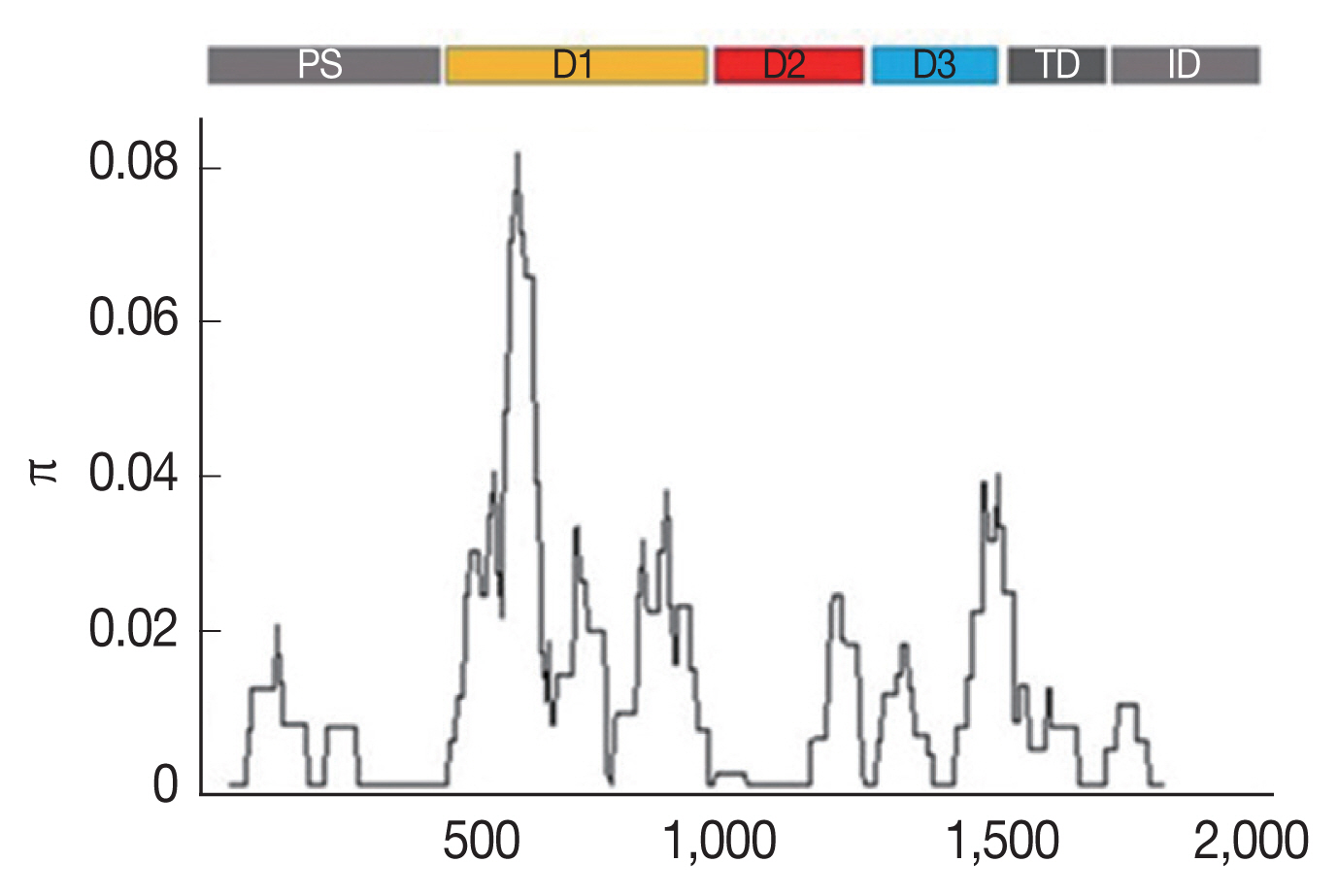
The SNPs were unevenly distributed across the AMA-1 gene. As shown in Supplementary Table S3, 34, 6, and 7 SNPs were located in domains I, II and III, respectively, whereas 2 and 1 SNPs were located in the flanking regions between domains I and II and between domains II and III, respectively. The other 10 SNPs were detected in the prosequence, the transmembrane domain, and the intracellular domain of AMA-1. The K-value for full-length AMA-1 in samples of Thailand was 20.798 (Table 1). The highest nucleotide difference was found at domain I (K=12.002), while the lowest was found at the transmembrane domain and intracellular domain (K=1.484). The analysis of the nucleotide sequence diversity (π) of domains I, II and III revealed π indices of 1.6449, 0.0058, and 0.1468, respectively, with an overall nucleotide sequence diversity (π) of AMA-1 of 0.01141 (Table 1; Fig. 1). Together, these results indicated that the most polymorphic region of the gene was in domain I.
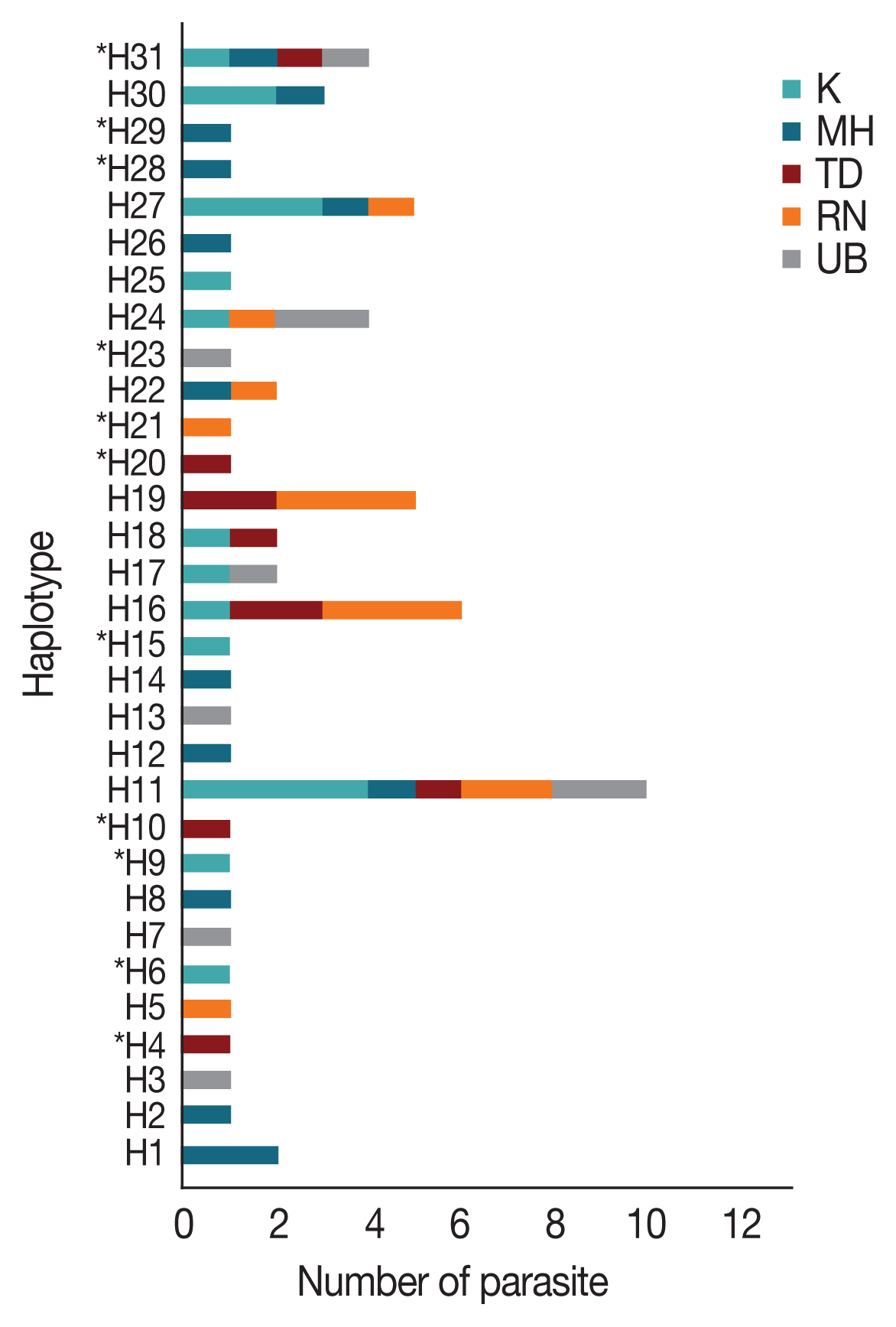
Multiple sequence alignment of the AMA-1 gene of 65 P. falciparum isolates also identified 31 unique haplotypes (H) of the AMA-1 sequences. The haplotype diversity index (Hd) of AMA-1 was estimated to be 0.952. Twenty-one AMA-1 haplotypes were detected in single localities. On the Thailand-Myanmar borders, 6, 6, and 2 haplotypes were detected in Mae Hong Son, Kanchanaburi and Ranong provinces, respectively. Additionally, 4 and 5 haplotypes were found only in Ubon Ratchatani and Trat, respectively, near the Thailand-Laos and Thailand-Cambodia borders, respectively. Of the 31 haplotypes identified, H11 was the most abundant and was distributed in all the sampled localities (Fig. 2). The haplotype analysis also showed that 13 and 3 of these haplotypes had previously been reported in P. falciparum populations in Thailand and Papua New Guinea, respectively [31,32,40] (Supplementary Table S4). The same AMA-1 haplotypes were also detected in parasites from Myanmar and 11 parasite isolates originated from Western Thailand (e.g. Mae Hong Son, Kanchanaburi and Ranong) [47]. In addition, 3 haplotypes were found to match the AMA-1 sequences of P. falciparum laboratory strains. Haplotypes H3 and H7 also matched the AMA-1 sequences of parasite isolates Dd2 and D10, respectively, from Indochina [32], while H19 corresponded to the sequence of the reference strain 3D7 [21]. As a result, the present study has identified 11 novel AMA-1 haplotypes that were unique to P. falciparum populations in Thailand (Fig. 2). When the AMA-1 sequence data from the present study (n=65 alleles) were combined with the full-length AMA-1 sequences from earlier studies (n=134 alleles) [31,32,40,42], a total of 47 unique haplotypes in 199 AMA-1 alleles were identified in the parasite populations in Thailand, with K, Hd and π values of 20.840, 0.9410, and 0.1128, respectively.
Evidence of natural selection of AMA-1
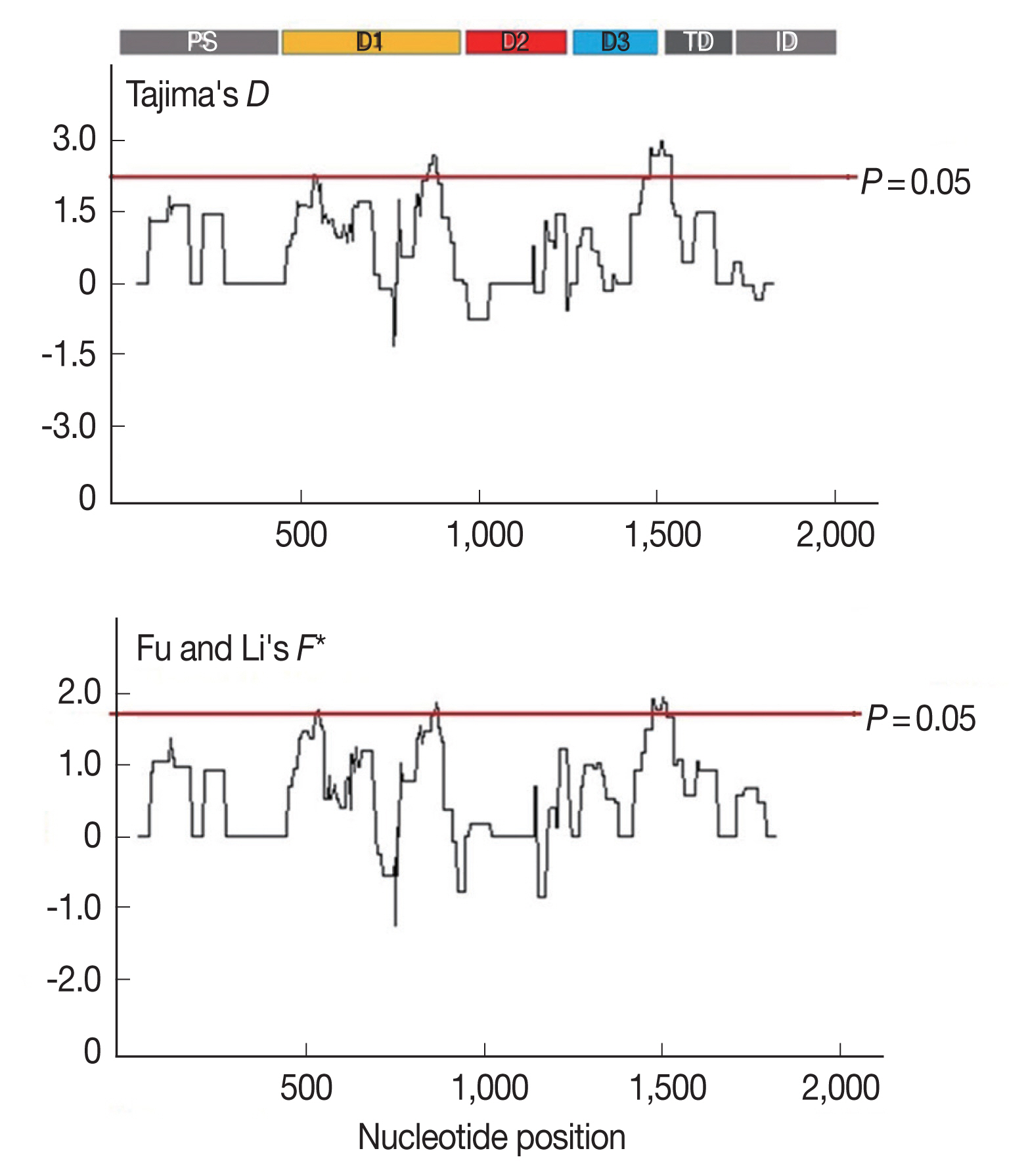
To identify the signature of natural selection in AMA-1, 3 within-population tests for neutrality (Tajima’s D, Fu and Li’s D* and Fu and Li’s F* tests) were performed using the 65 AMA-1 alleles in the present study. The Tajima’s D and Fu and Li’s F* values were significant, with values of 1.906 (P <0.05) and 1.6448 (P <0.05), respectively. However, although the Fu and Li’s D* value (D*=1.03967; >0.10) was positive it was not significant. When the AMA-1 sequences (134 alleles) in Thailand from previous studies [31,32,40,42] were combined with the 65 alleles of this study and analyzed, similar results were obtained with significant Tajima’s D and Fu and Li’s F* values of 2.200 (<0.05) and 2.0362 (P <0.05), respectively, while the Fu and Li’s D* value (D*=1.02317; P >0.10) remained positive but insignificant. These results indicated a tendency for positive diversifying selection in the AMA-1 gene. To further examine the possibility of natural selection, the dN/dS ratio was calculated using the sequences of the 65 alleles. The dN/dS ratio was 5.8211 (greater than 1). Taken together, the results of the neutrality tests and the dN/dS ratio supported the view that the positive (diversifying) selection may contribute to the diversity of the AMA-1 gene.
Furthermore, to determine whether specific regions of AMA-1 were under positive selection, a sliding window plot analysis was performed (Fig. 3). The values of D and F* were calculated based on a window of 100 bp moving in 3-bp steps. According to the Tajima’s D test, 3 regions of AMA-1 were found to be under positive selection: nucleotide positions 502–573 and 829–906 in domain I and 1,450–1,545 in domain III. Similar regions spanning nucleotide positions 502–585, 808–921, and 1,429–1,506 showed a significant departure from neutrality in the Fu and Li’s F* test. Thus, it is likely that positive diversifying selection mainly contributed to the genetic polymorphism in domains I and III of AMA-1.
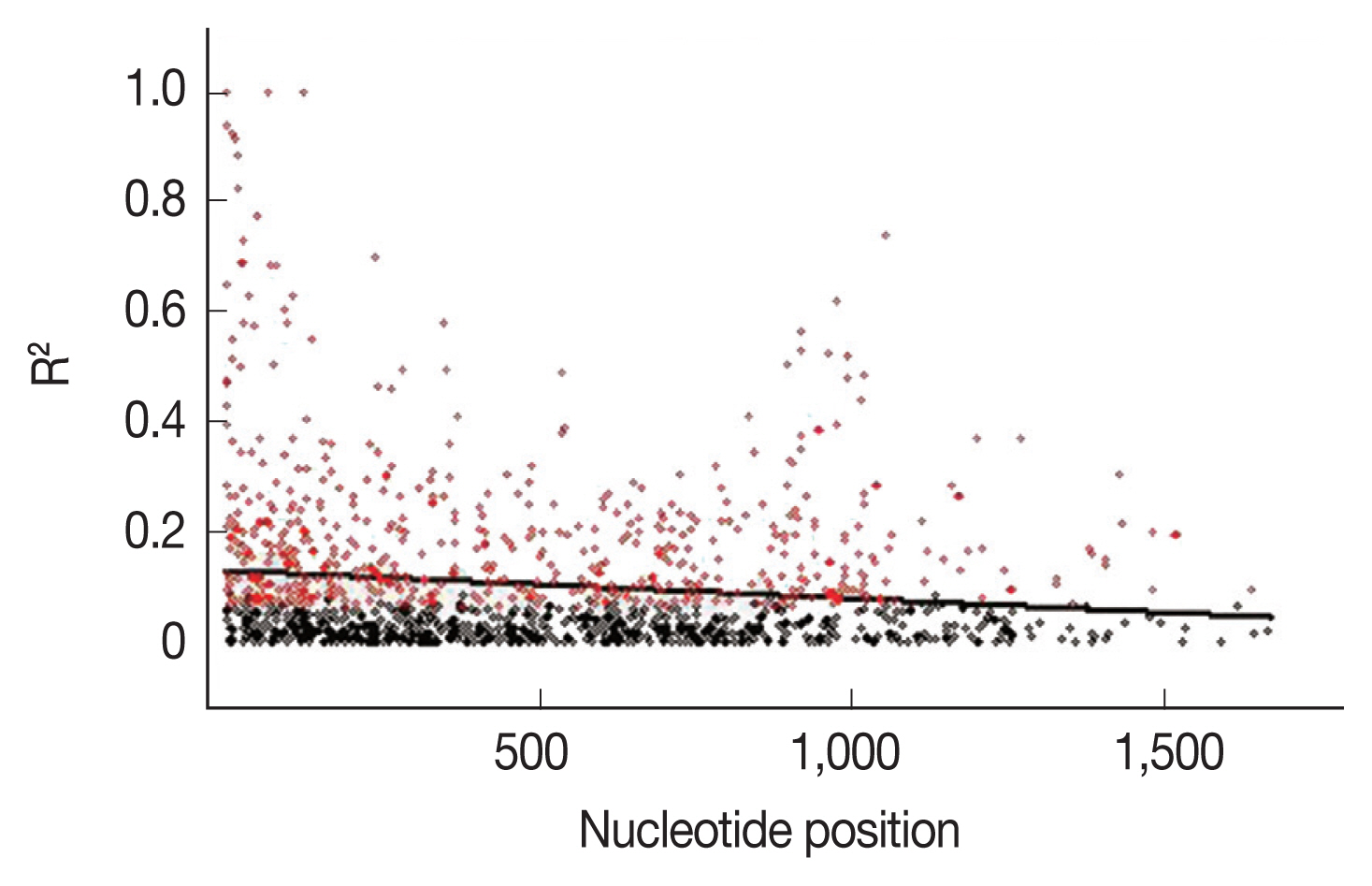
To further explain the high mutation rates at the AMA-1 locus, linkage disequilibrium was calculated for all the non-singleton polymorphic residues and plotted against the nucleotide distance. Fig. 4 demonstrated that the linkage disequilibrium index (R2) declined rapidly with increasing nucleotide distance. It can be assumed that a high meiotic recombination rate may contribute to the increased polymorphism in the AMA-1 locus. A minimum of 20 recombination events (Rm) was predicted to have occurred to give rise to the 31 haplotypes observed in these 65 alleles. The recombination parameter had high values of 0.0371 between adjacent nucleotide sites (Ra) and 68.6 for the whole sequence (Rb) in P. falciparum populations in Thailand. Overall, this result was consistent with the previous analysis of recombinant parameters using the partial AMA-1 sequences from P. falciparum populations from Western Thailand [45].
Population structure of P. falciparum based on the AMA-1 gene
The distribution of the 65 AMA-1 alleles in P. falciparum in Thailand (this study) also revealed different levels of genetic diversity in the different geographical populations. The highest genetic diversity of AMA-1 was detected in P. falciparum populations near the Thailand-Myanmar borders, with a total of 12 and 11 haplotypes being identified in Kanchanaburi and Mae Hong Son, respectively. On the other hand, the parasite population in Ubon Ratchatani had the lowest genetic diversity of AMA-1, with a total of 8 haplotypes being identified. To compare the patterns of distribution of AMA-1 haplotypes between P. falciparum populations in Thailand, pairwise inter-population comparisons were performed for each population using Wright’s Fixation index (Fst). Table 2 shows that a significant Fst value (P =0.03) was found between the parasite populations in Mae Hong Son and Ranong, indicating potential population sub-division between these 2 P. falciparum populations. In contrast, the Fst values from the other pairs of all 5 parasite populations were low and non-significant (P >0.05). When the previously reported AMA-1 haplotypes of P. falciparum in Tak, Western Thailand [31,42,45] were included in the Fst analysis, the Fst values remained low and non-significant between P. falciparum in Tak and the other populations in Thailand. Therefore, this result inferred that the P. falciparum parasite populations (or at least the AMA-1 gene) circulating in this region were genetically homogeneous.
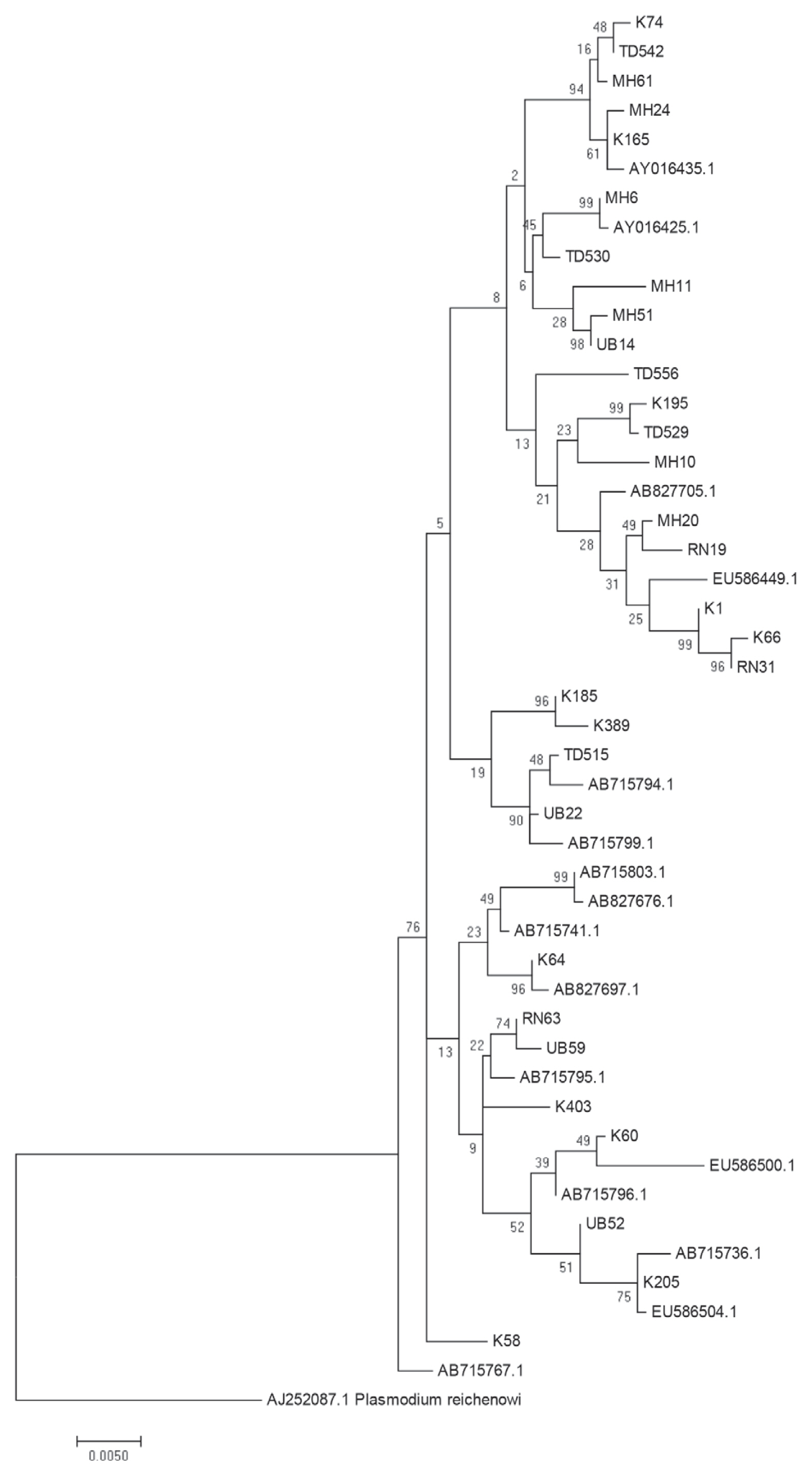
The above data provide suggestive evidence of the genetic flow between allopatric P. falciparum populations near the borders of Thailand with Myanmar, Laos and Cambodia. To further examine the close genetic relationship of the P. falciparum AMA-1 gene, ML-based phylogenetic trees of the 47 unique AMA-1 haplotypes were constructed, with the highest log likelihood tree shown in Fig. 5. The AMA-1 haplotypes formed 3 major clades, where the AMA-1 genes in each clade were from multiple geographical origins, reinforcing the idea that genetic recombination is very successful between closely related parasite populations.
Furthermore, sequences of AMA-1 domain I from P. falciparum populations in 16 countries worldwide, including 3 countries in each of Southeast Asia (Myanmar, the Philippines, and Malaysia), the Middle East and South Asia (India, Saudi Arabia, and Iran), and Oceania (The Solomon Islands, Vanuatu, and Papua New Guinea), 6 countries in Africa (The Gambia, Ghana, Tanzania, Nigeria, Kenya, and Mali) and one country in South America (Venezuela), were retrieved through a nucleotide database search at the NCBI website (Supplementary Table S2). A total of 3,311 alleles, including the 65 Thai sequences from the present study, were obtained. Pairwise inter-population comparisons were performed between the AMA-1 haplotypes of P. falciparum in Thailand and those of the other countries. The Fst values were significant between the P. falciparum population in Thailand and other populations, except for Myanmar (Table 2), suggesting a similar allelic distribution of the AMA-1 gene in the Greater Mekong Subregion (GMS). The analysis also inferred that population differentiation has occurred between P. falciparum in the GMS and other countries.
DISCUSSION
The present study aimed to investigate the extent of haplotype diversity in the P. falciparum AMA-1 gene in Thailand. The main finding was that the polymorphic P. falciparum AMA-1 gene within this region had as many as 31 haplotypes, which were characterized by 60 SNP loci across the gene. The SNPs were distributed mainly in the ectodomain of the AMA-1 gene, which is in agreement with previous reports that domain I of AMA-1 was the most polymorphic region [36,40,45]. Nearly 2/5 (41%) of the AMA-1 haplotypes identified in the present study had previously been detected in Tak, Western Thailand [31,32,40], while the level of nucleotide diversity of AMA-1 in P. falciparum in Thailand was similar to a previous report on P. falciparum populations in Myanmar [46]. Interestingly, 5 haplotypes (16%) of the AMA-1 gene identified in the present study were identical to those identified in P. falciparum in Myanmar [47], indicating the close genetic relatedness between P. falciparum populations in Thailand and China-Myanmar. This hypothesis was supported by Wright’s statistics, which suggests that gene flow operates among P. falciparum populations in the GMS. However, note that the P. falciparum samples from Southern China and Vietnam were not included in this study and should be included in further studies to generate a more complete view of the P. falciparum AMA-1 genetic diversity in Southeast Asia.
The Wright Fst analysis also indicated that the population structure of P. falciparum in Southeast Asia, based on the AMA-1 gene, was significantly different from the populations in the Middle East, South Asia, Oceania, Africa and South America. This result was similar to a separate study using the P. falciparum merozoite surface protein-3 gene [49,50]. The population subdivision among Asia, Africa and South America has also been reported using whole genome sequence data [5]. The different population structures, using either the AMA-1 gene or the whole genome sequences, reflect the different intensity and prevalence of malaria parasites in each geographical region. In Southeast Asian countries, the malaria transmission rate is low, seasonal and stable, while in Africa, malaria is holoendemic [43,47,60]. Despite the different population structures, the number of AMA-1 sequences shared among the parasite populations, however, suggests that similar selective forces act in the different geographical regions.
In addition to the population structure of the malaria parasite, sequence analysis of AMA-1 alleles also revealed that amino acid substitution is generally favored with a clear signature of positive selection in the AMA-1 locus among Thai parasite isolates. That is the number of non-synonymous substitutions per non-synonymous site significantly exceeded the number of synonymous substitutions per synonymous site [61]. This adaptive evolution is presumably due to host immune selection. Additionally, the results from neutrality tests further supported positive diversifying selection operating at the P. falciparum AMA-1 locus, while the window analysis of Tajima’s D and Fu and Li’s D* tests suggested that positive selection acts specifically on domains I and III. Although the values of Fu and Li’s F* value test were not significant across the entire region, the positive values indicated the tendency of departure from neutral evolution and towards positive diversifying selection. This result concurs with the finding from previous studies in P. falciparum in Nigerian and Thai populations, which showed that excessive polymorphism in domains I and III of AMA-1 was maintained by positive selection, while no evidence of selection was detected for domain II [33,36,45]. In addition, there is evidence that the polymorphic sites in domains I and III are targets of inhibitory antibodies [62–64]. Taken together, these results imply that domains I and III are potential targets for protective immunity and could be used as vaccine subunits.
Current AMA-1 vaccines that are being tested in Phase I and II clinical trials are based on the sequence of P. falciparum strain 3D7 and/or FVO [19,22,65]. In the present study, only one (H19) of the 31 AMA-1 haplotypes matched the 3D7 strain. A previous analysis of the 135 AMA-1 alleles from P. falciparum in Myanmar showed that none of them was identical to the 3D7 strain, although a few isolates had the AMA-1 sequence of the FVO strain [46]. Accordingly, it is postulated that monovalent or bivalent vaccines may offer little protection to the immunized subjects in the GMS. It has been shown using the rodent malaria model that the AMA-1 vaccine elicited immune protection against parasites with the homologous AMA-1 allele but not those with a heterologous allele [16]. Likewise, the results from Phase II clinical trials showed that vaccinations with a monovalent AMA-1 vaccine (FMP2.1/AS) in children in Mali conferred 20% efficacy that was strain-specific [66]. Nonetheless, the analysis of global AMA-1 haplotypes showed that several major alleles of AMA-1 were shared between endemic areas regardless of geographical location [32]. The inclusion of multiple P. falciparum AMA-1 alleles as vaccines according to the local population structure of P. falciparum may give a better protective efficacy of AMA-1 if it is allele-specific [67]. Hence, there is no doubt that estimates of molecular diversity of the major vaccine candidate, AMA-1, have practical implications for the rational design of effective AMA-1-based vaccines.
In conclusion, we have formed an initial AMA-1 sequence database of P. falciparum in Thailand comprised of baseline data of P. falciparum AMA-1 polymorphisms in natural isolates that originated from the national borders near Myanmar, Laos and Cambodia. Novel haplotypes of AMA-1 were found and strongly support the presence of positive selection acting on the gene. Because countries in Southeast Asia are entering the age of ASEAN Economic Community (AEC), with an increased movement of people between the countries, the continuous introduction and exposure of imported malaria cases is expected to increase. This would potentially contribute to an increased level of genetic recombination and polymorphism of the malaria parasite within and between populations in the AEC region, presenting new challenges to malaria elimination programs. An understanding of the genetic polymorphism in any vaccine candidate, including AMA-1, is necessary for malaria vaccine development in light of the extensive antigenic diversity and allele-specific immunity.