Proliferation of Toxoplasma gondii Suppresses Host Cell Autophagy
Article information
Abstract
Autophagy is a process of cytoplasmic degradation of endogenous proteins and organelles. Although its primary role is protective, it can also contribute to cell death. Recently, autophagy was found to play a role in the activation of host defense against intracellular pathogens. The aims of our study was to investigate whether host cell autophagy influences Toxoplasma gondii proliferation and whether autophagy inhibitors modulate cell survival. HeLa cells were infected with T. gondii with and without rapamycin treatment to induce autophagy. Lactate dehydrogenase assays showed that cell death was extensive at 36-48 hr after infection in cells treated with T. gondii with or without rapamycin. The autophagic markers, LC3 II and Beclin 1, were strongly expressed at 18-24 hr after exposure as shown by Western blotting and RT-PCR. However, the subsequent T. gondii proliferation suppressed autophagy at 36 hr post-infection. Pre-treatment with the autophagy inhibitor, 3-methyladenine (3-MA), down-regulated LC3 II and Beclin 1. The latter was also down-regulated by calpeptin, a calpain inhibitor. Monodansyl cadaverine (MDC) staining detected numerous autophagic vacuoles (AVs) at 18 hr post-infection. Ultrastructural observations showed T. gondii proliferation in parasitophorous vacuoles (PVs) coinciding with a decline in the numbers of AVs by 18 hr. FACS analysis failed to confirm the presence of cell apoptosis after exposure to T. gondii and rapamycin. We concluded that T. gondii proliferation may inhibit host cell autophagy and has an impact on cell survival.
INTRODUCTION
Autophagy promotes cell survival and cellular homeostasis by removing any damaged elements from the cells [1,2]. It can also lead to type II programmed cell death (PCD) which is distinct from apoptosis (type I PCD). Unlike apoptosis, type II PCD is independent of caspase activation and is characterized by the presence of autophagic cytoplasmic vacuoles containing sequestered cytoplasm and organelles. These autophagosomes later fuse with lysosomes and their contents are degraded [3-6]. Autophagy can lead to cell survival and cell death, and factors affecting these outcomes are poorly understood.
Toxoplasma gondii (T. gondii) is an intracellular protozoan parasite that replicates within the parasitophorous vacuole (PV) protected from lysosomal fusion. T. gondii is able to exploit host cell autophagy for its own nutrition [7]. Yet, the relationship between T. gondii proliferation and host cell autophagy is unclear. There are 2 pathways acting up-stream of autophagy; PI3K and Akt. Activation of class I PI3K inhibits autophagy through the activation of protein kinase B (Akt) and mTOR. By contrast, class III PI3K in a complex with Beclin 1 promotes autophagy [8,9]. Rapamycin is an antibiotic and immunosuppressant that inhibits the activity of mTOR pathway and enhances autophagy [10]. To detect autophagy, microtubule-associated protein 1 light chain 3 (LC3) is used. LC3 I (18 kDa) and its proteolytic product, LC3 II (16 kDa) are localized in the cytoplasm and on autophagosomal membranes, respectively. Under conditions of autophagy, LC3 I is converted into LC3 II.
The formation of autophagic vacuoles is thought to be induced by Beclin 1 (Atg6), one of the proteins required for the autophagic process [11,12]. It has been found to promote cell survival and it may function as a tumor suppressor in specific conditions [13]. Autophagic vacuoles were detected by monodansyl cadaverine (MDC) staining and transmission electron microscopy (TEM). In this experiment, we found that cell autophagy is inhibited by T. gondii proliferation.
MATERIALS AND METHODS
Toxoplasma gondii and HeLa cell culture
T. gondii (RH strain) tachyzoites were maintained by intraperitoneal infection of ICR mice (Osan, Kyunggi-do, Korea) at intervals of 3 or 4 days. The tachyzoites were harvested from the mice with 5 ml RPMI 1640 medium, washed with PBS and centrifuged at low and high speeds (500 g and 3,000 g) for 5 min to remove the peritoneal cells. HeLa cells (human cervical carcinoma, from Yonsei University) were cultured in RPMI 1640 with 10% FBS, penicillin (10 U/ml) and streptomycin (0.1 mg/ml) at 37℃ and 5% CO2.
Reagents
The assays and reagents were sourced as follows: CytoTox96® non-radioactive cytotoxicity assay (Promega, Madison, Wisconsin, USA); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Roche, Manheim, Germany); RPMI 1640 (Welgene, Gyeongsan-si, Gyeongsangbuk-do, Korea); rapamycin, MDC, LC3 antibody, 3-methyladenine (3-MA), penicillin and streptomycin (Sigma Aldrich, St. Louis, Missouri, USA); Beclin-1 antibody (Santa-Cruz Biotechnology, Santa-Cruz, California, USA); β-actin antibody (Cell Signaling, Beverly, Massachusetts, USA); z-VAD-fmk (EMB Biosciences, Darmstadt, Germany), calpeptin (Calbiochem, La Jolla, Califormia, USA); Lammli sample buffer (Bio-Rad, Hercules, California, USA); QIAzol (QIAgen, Dusseldoef, Germany); the amfiRiver 1-step RT-PCR kit (GenDepot, Barker, Texas, USA); and TRIzol reagent (Invitrogen, Carlsbad, California, USA).
T. gondii infection and induction of autophagy
HeLa cells (1×106/ml) were seeded in 24-well culture plates with round cover glasses and exposed to T. gondii (cell: tachyzoite ratio=1:5) or rapamycin (1 µM) to induce autophagy or to both T. gondii and rapamycin. The cells were incubated in RPMI 1640 with 10% FBS and sampled after 6, 18, 24, 36, and 48 hr at 37℃ in 5% CO2. The relationship between autophagy and apoptosis was assessed by pre-treatment of the HeLa cells for 2 hr with an inhibitor of autophagy, 3-MA (10 nM), the pan-caspase inhibitor, z-VAD-fmk (20 µM), and the calpain inhibitor, calpeptin (100 µM) [14].
Lactate dehydrogenase (LDH) and MTT assays
HeLa cells (1×105/100 µl) were cultured overnight in 96-well plates containing RPMI 1640 with 10% FBS at 37℃ and 5% CO2. CytoTox96® non-radioactive cytotoxicity assays were performed to assess cell death by measuring the release of lactate dehydrogenase (LDH) into the medium by the T. gondii-infected cells. Culture supernatants were collected and transferred to another 96-well plate and 50 µl of the substrate mixture was added. After incubation at room temperature for 30 min in the dark, 50 µl of a stop solution was added to the wells. After 1 hr, optical density (OD) values at 490 nm were read on an ELISA reader (Molecular Devices, California, USA) [12]. To measure the cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was performed with the Cell Proliferation Kit (Roche). Briefly, HeLa cells (1×105 cells/well) were cultured overnight in 96-well plates in RPMI 1640 with 10% FBS at 37℃ and 5% CO2, and T. gondii (5×105) and rapamycin (1 µM) were added for 6, 24, 36, and 48 hr. A reagent buffer (10 µl) was added and incubation continued for 4 hr, followed by the addition of solubilization solution (100 µl) and incubation overnight at 37℃, 5% CO2. Absorbance was measured at 570 nm on the ELISA reader [15].
Western blotting
Protein concentrations were determined by the Bradford method (Bio-Rad). HeLa cells (1×106) were cultured overnight in 24-well culture plates containing RPMI 1640 with 10% FBS and exposed to T. gondii (5×106) and/or rapamycin (1 µM) for 6, 18, 24, or 36 hr. Levels of LC3 and Beclin 1 were determined by Western blotting. Briefly, cell lysates were fractionated by 12% SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad) using a semi-dry electro-transferring unit. After blocking with 5% skim milk for 1 hr at 37℃, the membranes were incubated with anti-LC3 Ab (1:1,000), anti-Beclin 1 Ab (1:1,000) or anti-β actin Ab (1:1,000) as primary antibodies (Abs) overnight at 4℃, followed by horseradish peroxidase conjugated anti-rabbit IgG polyclonal Ab as the secondary Ab (1:2,000) (Assay Designs, Ann Arbor, Michigan, USA) for 1 hr at room temperature. After washing, protein bands were detected using the PLUS Chemiluminescence Detection System (Amersham Pharmacia Biotech., Piscataway, New Jersey, USA).
RT-PCR
HeLa cells (1×106) were cultured in 12-well culture plates overnight at 37℃ and 5% CO2 and incubated with T. gondii (5×106) and/or rapamycin (1 µM) for 1, 18, 24, or 48 hr. Total RNA was extracted with TRIzol (Invitrogen) and 200 µl chloroform (Sigma-Aldrich). The cDNAs were synthesized according to the manufacturer's instructions using the 1-step RT-PCR kit. Reverse-transcription was performed in a total volume of 20 µl (1 µg of total RNA) and cDNA was amplified with Beclin 1 and β-actin primers. For Beclin 1, the conditions were as follows: 94℃ for 10 min, 94℃ for 20 sec, 54℃ for 30 sec, 72℃ for 40 sec, 45 cycles. For β-actin, the conditions were as follows: 95℃ for 10 min, 95℃ for 45 sec, 58℃ for 2.5 min, 72℃ for 30 sec, 30 cycles. The following primer sequences were used for amplification.
(F) AGGAACTCACAGCTCATTAC, (R) AATGGCTCCTCTCCTGAGTT for Beclin 1;
(F) CCAGAGCAAGAGAGGTATCC, (R) CTGTGGTGGTGAAFCTGTAG for β-actin.
MDC staining
HeLa cells (1×106) were cultured in 12-well culture plates on cover glasses for 6 hr at 37℃, 5% CO2 in RPMI 1640 with 10% FBS. The cells were infected with T. gondii (cell: tachyzoite ratio=1:5) and/or rapamycin and incubated for 6, 18, and 24 hr. A stock solution of 10 mM MDC, the autofluorescent dye, was prepared in DMSO and the cells were incubated with 0.05 mM MDC at 37℃, 5% CO2 for 1 hr. After washing a fixation-permeabilization solution (Becton Dickinson, San Jose, California, USA) was added for 15 min [16]. After fixing, the cells were washed 3 times with a permeabilization-washing buffer, transferred to glass-slides and observed under a fluorescence microscope (DMRE, Leica, Germany).
Transmission electron microscopy (TEM)
HeLa cells (5×105) were incubated in 24-well plates overnight and treated with T. gondii and/or rapamycin (1 µM) for 6, 18, 24, or 48 hr at 37℃, in a CO2 incubator with or without 3-MA or wortmannin. The cells were pre-fixed in a solution of 3% glutaraldehyde in 0.1 M PBS (pH 7.8) for 3 hr at room temperature and post-fixed in 1% osmium tetroxide for 1.5 hr at 4℃. The samples were dehydrated in increasing concentrations of ethanol (50%, 70%, and 100%) and embedded in Epon resin and ultrathin sections were loaded on copper grids. They were stained with uranyl acetate and lead citrate, and examined with a transmission electron microscope (Hitachi, H-7600s, Tokyo, Japan).
Flow cytometry
HeLa cells (1×106) were dispensed onto 6-well culture plates and incubated in RPMI 1640 with 10% FBS and actinomycin D (5 µg/ml), rapamycin (1 µM), and T. gondii for 18 hr at 37℃ and 5% CO2. The cells were collected, washed, and fixed in 70% ethanol at 4℃ for 18 hr. After washing with PBS, they were treated according to the manufacturer's protocol of the apoptosis detection kit (Becton Dickinson). The cells were suspended in binding (200 µl) buffer and transferred to a FACS tube. Annexin-V (5 µl) and propidium iodide (PI) (5 µl) were added and after mixing, the tubes were kept in the dark at room temperature for 15 min. The cells were then suspended in binding buffer (400 µl) for 1 hr and analyzed with a FACSCalibur™ flowcytometer (Becton Dickinson) [9].
Statistical analysis
Data are presented as mean±SD. Statistical significance was determined with Student's t-tests. P-values of <0.05 were considered statistically significant.
RESULTS
Cell death after T. gondii infection and rapamycin treatment
The LDH and MTT assays were performed to investigate the role of autophagy in cell death and survival after exposure to T. gondii and rapamycin. Low levels of cell death were observed at 24 hr post-infection which subsequently increased at 36 hr and 48 hr (control 18%, P<0.05; Fig. 1A). In both assays, the control cells were treated with rapamycin for 24 hr. There was no difference in the rates of cell death between infected cells with and without rapamycin treatment. Conversely, cell viability decreased gradually, as observed at 24 hr and 48 hr post-infection (control 80%, P<0.05; Fig. 1B).
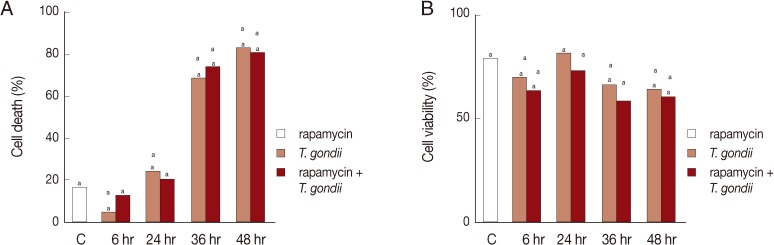
Cell death and cell viability were assessed by the LDH and MTT assay. HeLa cells were exposed to T. gondii or rapamycin or combination for 6-48 hr. Control cells were treated with rapamycin only for 24 hr. (A) Cell death increased due to T. gondii or T. gondii plus rapamycin treatment at 36 hr and 48 hr post-infction as shown by the LDH assay. (B) The MTT assay showed gradually decreasing cell viability following exposure to T. gondii or T. gondii plus rapamycin after 24 hr. C, control. aP<0.05.
Induction of autophagy by T. gondii and rapamycin
LC3 II and Beclin 1, the markers of autophagic vacuoles, were detected by Western blotting or RT-PCR. Cells exposed to either rapamycin or T. gondii were cultured as controls. LC3 II (18 kDa) and Beclin 1 (60 kDa) were detected 6 hr after treatment with a subsequent increase detected at 18 hr and 24 hr post-infection; however, they subsequently declined at 36 hr (Figs. 2A, 3A). Similarly, Beclin 1 mRNA was expressed strongly at 18 hr, only to decline again by 24 hr and 48 hr (Fig. 3C). Cells with or without rapamycin were cultured as controls. Next, we investigated the effects of autophagy inhibitors on apoptosis. Pre-treatment with 3-MA prior to T. gondii and rapamycin treatment suppressed the levels of LC3 II and Beclin 1. Cells pre-treated with z-VAD-fmk showed increased levels of the autophagy marker Beclin 1, but there was no increase in LC3 II (Figs. 2B, 3B). In turn, the levels of Beclin 1 decreased in the cells exposed to T. gondii and rapamycin after treatment with calpeptin (Fig. 3B).
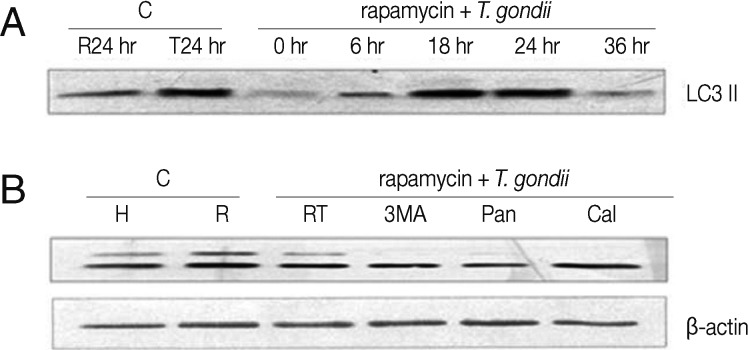
Autophagic marker LC3 II, measured by Western blotting. HeLa cells were exposed to T. gondii (cell: T. gondii ratio=1:5) or rapamycin (24 hr) or the combination for 6-36 hr. Cells were pre-treated with an autophagic inhibitor, 3-MA (10 nM), a pan-caspase inhibitor (20 µM) or a calpain inhibitor (100 µM) for 2 hr. (A) Over-expression of LC3 II was observed in T. gondii-infected HeLa cells with rapamycin treatment at 18-24 hr post-infection. (B) The 3-MA and pan-caspase inhibitor suppressed LC3 II expressison. C, control; H, HeLa cells; R, rapamycin; T, T. gondii; RT, rapamycin and T. gondii; 3MA, 3- methyladenine; Pan, pan-caspase inhibitor; Cal, calpain inhibitor.
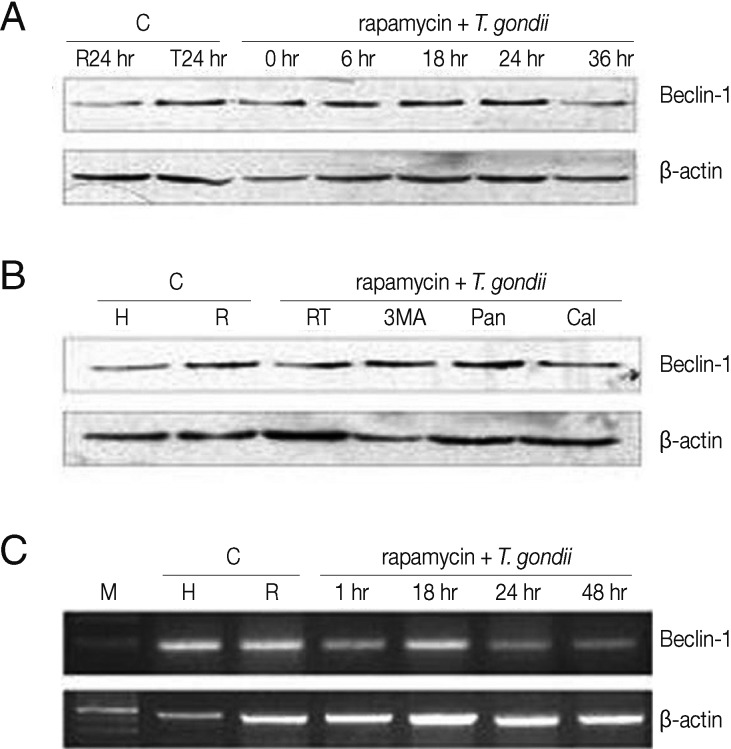
Beclin 1 expression in HeLa cells treated with T. gondii and rapamycin detected by Western blotting and RT-PCR. HeLa cells were exposed to T. gondii (cell: T. gondii ratio=1:5) or rapamycin or the combination for 1-48 hr or for 6-36 hr. Cells were pre-treated with 3-MA (10 nM), pan-caspase inhibitor (20 µM) or a calpain inhibitor (100 µM) for 2 hr followed by T. gondii and rapamycin treatment. (A and C) Increased expression of Beclin 1 mRNA and protein was observed in HeLa cells treated with T. gondii or T. gondii plus rapamycin at 18 hr and 24 hr post-infection. (B) Beclin 1 expression was suppressed in cells pre-treated with 3-MA and a calpain inhibitor for 2 hr. C, control; H, HeLa cells; R, rapamycin; T, T. gondii; RT, rapamycin and T. gondii; 3-MA, 3-methladenine; Pan, pan-caspase inhibitor; Cal, calpain inhibitor.
MDC positive autophagic vacuoles
Fluorescence microscopy detected MDC accumulation in the autophagic vacuoles. Distinct fluorescent dot-like structures corresponding to AVs were observed in the positive control cells treated with rapamycin for 18 hr, while the cells without rapamycin treatment showed no fluorescence (Fig. 4A, B). MDC-positive AVs were also detected in the cells exposed to T. gondii and rapamycin and their levels peaked at 18 hr and their fluorescent density had decreased by 24 hr post-infection (Fig. 4C, D).
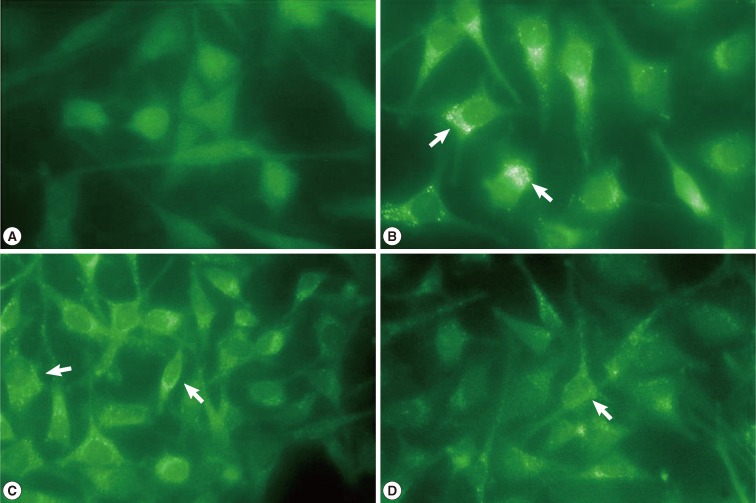
Fluorescence micrographs of MDC-labeled autophagic vesicles (AVs). HeLa cells were exposed to T. gondii (cell: T. gondii ratio= 1:5) and rapamycin for 6, 18, and 24 hr. The cells were incubated with MDC (0.05 mM) for 1 hr and treated with a fixation-permeabilization solution for 15 min. Untreated cells or cells treated with rapamycin for 18 hr were used as controls (A, B; ×400). Distinct fluorescent dots were observed in the cytoplasm of the cells infected with T. gondii and exposed to rapamycin for 18 hr. Decreased fluorescence was noted at 24 hr post-infection (C, D; × 200). Arrows indicate the MDC positive AVs.
T. gondii proliferation in autophagic cells observed by TEM
To confirm autophagy, the cells infected with T. gondii and exposed to rapamycin were observed by TEM. Many autophagic vacuoles containing assorted organelles appeared in the cytoplasm of controls treated with rapamycin for 18 hr, and T. gondii proliferation in PVs was observed at 18 hr post-infection (Fig. 5A, B). Large numbers of autophagic vacuoles were visualized at 6 hr post-treatment in the T. gondii-infected, rapamycin-treated cells (Fig. 5C). However, the subsequent tachyzoite proliferation in PVs coincided with a decline in the numbers of AVs at 18 hr post-infection (Fig. 5D). Pre-treatment with the autophagy inhibitor, 3-MA for 2 hr significantly reduced the numbers of AVs, but the same effect was not seen after pre-treatment with wortmannin (Fig. 5E, F).
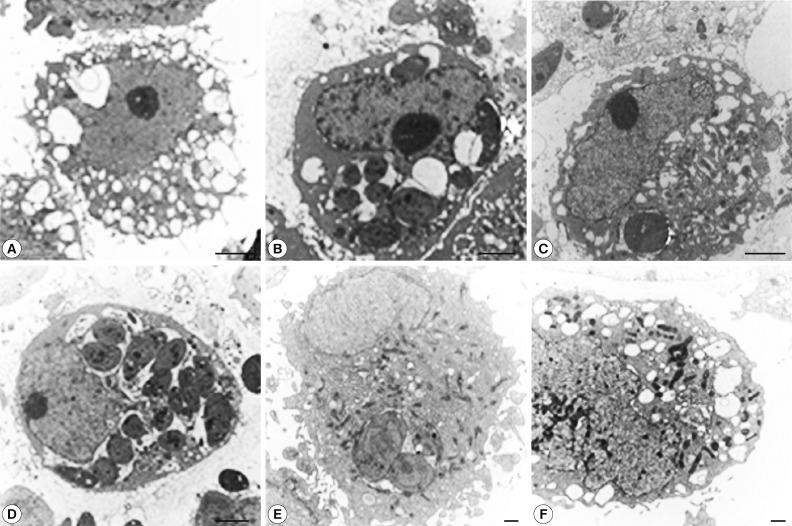
Ultrastructural observation of T. gondii-infected and rapamycin-treated HeLa cells. (A) Control cells treated with rapamycin for 18 hr showing numerous AVs containing damaged organelles. (B) Cells infected with T. gondii for 18 hr showing tachyzoite proliferation in PVs. (C) HeLa cells exposed to T. gondii and rapamycin showing numerous AVs at 6 hr post-infection. (D) Proliferation of T. gondii accompanied by a decline in the numbers of Avs, observed at 18 hr post-infection. (E, F) Cells pre-treated 3-MA for 2 hr and exposed to T. gondii with rapamycin for 18 hr had reduced numbers of AVs. Cells pre-treated with wortmannin did not show this effect. Scale bar: 2 µm (A-D), 500 nm (E, F).
Absence of apoptosis in the cells exposed to T. gondii and rapamycin
Cell death due to T. gondii infection and rapamycin treatment was investigated by flow cytometry after annexin V/ PI double staining. In positive controls, treated with actinomycin D (5 µg) for 18 hr, the rate of apoptosis reached 58.3%, and in the untreated HeLa cells, apoptosis reached 14.7% (Fig. 6Aa, b). Conversely, rapamycin treatment seemed to suppress apoptosis (10.1%), as did exposure to both T. gondii and rapamycin for 18 hr (13.4%; Fig. 6Ac, d). In the cells pre-treated with 3-MA, apoptosis reached 15.9% (Fig. 6Be), while pre-treatment with zVAD-fmk and calpeptin showed the rates of 13.7% and 16.0%, respectively (Fig. 6Bf, g). These findings suggested that simultaneous treatment with T. gondii and rapamycin resulted in the absence of apoptosis.
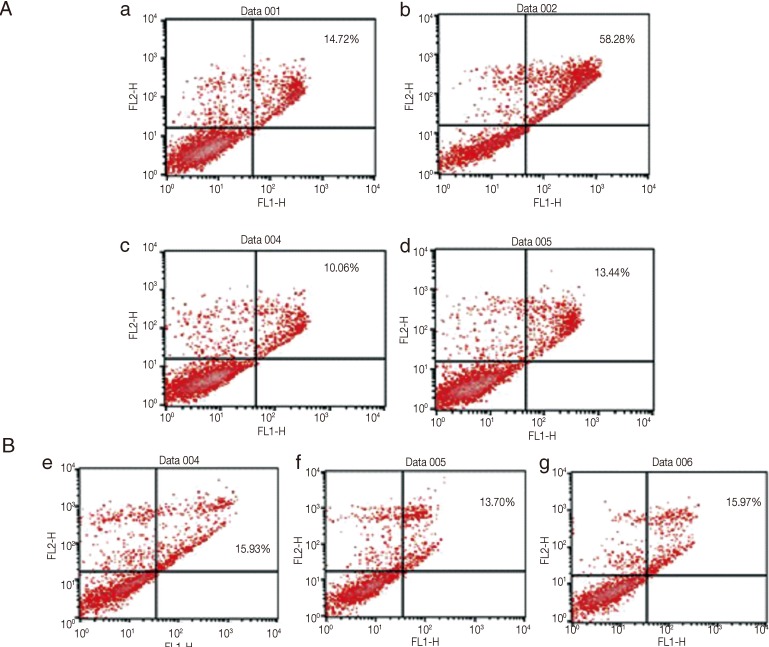
Flow cytometry to detect apoptosis in HeLa cells treated with T. gondii and rapamycin. (Aa, b) Control cells with no treatment or actinomycin D (5 µg/ml) had apoptosis rates of 14.7 % and 58.3%, respectively. (Ac, d) Low level apoptosis was observed in HeLa cells treated with rapamycin with or without T. gondii infection (13.4% and 10.1%, respectively). (Be, f, g) Cells pre-treated with inhibitors of autophagy or apoptosis and exposed to T. gondii and rapamycin did not show apoptosis (15.9% in cells pre-treated with 3-MA; 13.7% in cells pre-treated with a pan-caspase inhibitor; 16.0% in cells pre-treated with calpeptin).
DISCUSSION
Autophagy is a temporary survival mechanism that provides an alternative source of energy, and its inhibition accelerates cell death [17]. It is also known to contribute to the clearance of pathogens and the activation of host cell defense mechanisms. However, whether autophagy promotes cell survival or induces cell death is controversial [1,9]. T. gondii is an intracellular parasite whose proliferation is dependent on host cell autophagy [7,18]. However, the specific role of autophagy in T. gondii infection is not well understood. In our experiments, LDH assays revealed extensive cell death among the HeLa cells treated with T. gondii and rapamycin at 36 hr post-inoculation (Fig. 1).
Microtubule-associated protein 1 light chain 3 (LC3) has 2 forms. In the autophagic process, LC3 I, a soluble form in the cytoplasm, transforms into LC3 II which localizes on the autophagosome membrane [19]. The rate of this conversion was shown to increase after T. gondii infection [20]. Another protein with a known role in mediating autophagy is Beclin 1 [13]. Its increased expression has been found to promote autophagy as well as inhibit the cell's tumor forming potential. In T. gondii-infected cells, Beclin 1 was found to concentrate near the parasitophorous vacuoles [2,7]. Here, we observed that the levels of both LC3 II and Beclin 1 increased in the T. gondii- and rapamycin-treated HeLa cells at 18 hr and 24 hr, respectively, but decreased again by 36 hr (Figs. 2, 3). This may imply that host cell autophagy is inhibited by T. gondii proliferation. Autophagy can promote cell survival by suppressing apoptosis, but the relationship between autophagy and apoptotic cell death is not well understood [21,22].
Treatment with an autophagy inhibitor, 3-MA alone and in combination with z-VAD-fmk was previously shown to provide significant protection against cell death following amino acid starvation [12]. The 3-MA blocks the formation of the autophagosome, while z-VAD-fmk inhibits caspase and converts apoptosis into autophagy or other types of cell death [15,16,21]. Equally important, although more diverse in its impact on apoptosis, cell cycling and autophagy, is calpain a calcium-dependent cysteine protease widely distributed in the cytoplasm [15]. Here, we examined the relationship between autophagy and apoptosis by pre-treating the cells for 2 hr with 3-MA and the apoptosis inhibitors, z-VAD-fmk and calpeptin.
The 3-MA pre-treated cells showed down-regulated expression of LC3 II and Beclin 1. Conversely, treatment with z-VAD-fmk appeared to up-regulate Beclin 1, while calpeptin increased the expression of LC 3 II (Figs. 2, 3). These findings support the conclusion that autophagy was enhanced due to the inhibition of apoptosis. To assess the inducton of autophagy, we examined the accumulation of MDC-labeled vacuoles by fluorescence microscopy [19,23]. The vacuole clusters were distinct at 18 hr, but their numbers then decreased at 24 hr post-infection, which coincided with the appearance of the autophagy markers, LC3 II and Beclin 1 (Figs. 2-4). Previously, incorporation of MDC into vacuoles was found to be inhibited by 3-MA and wortmannin in cells under starvation conditions [14].
On TEM, numerous AVs were evident in the cytoplasm after 18 hr of rapamycin treatment as the control. It appears that, at 6 hr post-infection, the PVs containing tachyzoites and the AVs co-existed in the T. gondii-infected, rapamycin-treated cells. However, by 18 hr, the tachyzoites had begun to proliferate and the numbers of AVs had decreased (Fig. 5A, C, D).
Furthermore, we found that autophagic vacuolization was suppressed by pre-treatment with 3-MA, but not by wortmannin (Fig. 5E, F). Based on these findings, we propose that T. gondii proliferation may suppress host cell autophagy. Apoptosis of HeLa cells treated with T. gondii and rapamycin was assessed by flow cytometry after staining with annexin V-FITC and PI. In control cells, apoptosis was efficiently induced by actinomycin D (Fig. 6Ab). Only very low levels of apoptotic cell death were observed at 18 hr post-infection which indicates that autophagy and apoptosis do not co-exist in HeLa cells treated wth T. gondii and rapamycin (Fig. 6Ad). In line with this, we previously observed that host cell apoptosis induced by actinomycin D was inhibited by T. gondii infection [24].
Recent investigations have focused on the cross-talk between the apoptotic and autophagic signaling pathways [21]. Autophagy can be a precursor and even an initiator of apoptosis, and conversely, apoptotic signaling can lead to the activation of autophagy [12,25]. An example of this mechanism can be seen in human amniotic epithelial cells, where a link between autophagic cell death and apoptosis has been demonstrated [4]. Suppression of Beclin 1 in mammalian cells was shown to impair autophagy and sensitize the cells to apoptosis [13]. However, essential for the anti-apoptotic effect of Beclin 1 is its interaction with Bcl-2 or Bcl-xL [17,21,26,27].
In conclusion, autophagy is initiated in order to prolong cell survival, but when taken to extremes, it may lead to cell death [10]. Our findings suggest that autophagy is a cell survival mechanism in T. gondii- and rapamycin-treated HeLa cells soon after inoculation. However, the subsequent proliferation of the tachyzoites may suppress autophagic pathways in the host cells.
ACKNOWLEDGMENT
This work was supported by National Research Foundation of Korea Grant funded by the Korean Government (KRF-2008-314-E0070-7644).