Degradation of the Transcription Factors NF-κB, STAT3, and STAT5 Is Involved in Entamoeba histolytica-Induced Cell Death in Caco-2 Colonic Epithelial Cells
Article information
Abstract
Entamoeba histolytica is a tissue-invasive protozoan parasite causing dysentery in humans. During infection of colonic tissues, amoebic trophozoites are able to kill host cells via apoptosis or necrosis, both of which trigger IL-8-mediated acute inflammatory responses. However, the signaling pathways involved in host cell death induced by E. histolytica have not yet been fully defined. In this study, we examined whether calpain plays a role in the cleavage of pro-survival transcription factors during cell death of colonic epithelial cells, induced by live E. histolytica trophozoites. Incubation with amoebic trophozoites induced activation of m-calpain in a time- and dose-dependent manner. Moreover, incubation with amoebae resulted in marked degradation of STAT proteins (STAT3 and STAT5) and NF-κB (p65) in Caco-2 cells. However, IκB, an inhibitor of NF-κB, was not cleaved in Caco-2 cells following adherence of E. histolytica. Entamoeba-induced cleavage of STAT proteins and NF-κB was partially inhibited by pretreatment of cells with a cell-permeable calpain inhibitor, calpeptin. In contrast, E. histolytica did not induce cleavage of caspase-3 in Caco-2 cells. Furthermore, pretreatment of Caco-2 cells with a calpain inhibitor, calpeptin (but not the pan-caspase inhibitor, z-VAD-fmk) or m-calpain siRNA partially reduced Entamoeba-induced DNA fragmentation in Caco-2 cells. These results suggest that calpain plays an important role in E. histolytica-induced degradation of NF-κB and STATs in colonic epithelial cells, which ultimately accelerates cell death.
INTRODUCTION
Entamoeba histolytica, a tissue-invasive extracellular protozoan parasite, is the etiologic agent of human amoebiasis, causing colitis and liver abscess [1]. During infection, E. histolytica trophozoites attach to the colonic mucin layer, which can lead to destruction of the mucin layer by amoeba-secreted cysteine proteases and, ultimately, the induction of cell death in colonic epithelial cells in a contact-dependent manner [2]. Amoeba-induced host cell death in colonic tissues is closely linked to the provocation of tissue inflammation, mediated by IL-1β [3]. In addition, Gal/GalNAc lectin, an immunologic surface molecule expressed on the plasma membrane of amoebae, is important for their adherence to host cells in vitro and their subsequent death [4,5]. Various intracellular signaling molecules have also been identified that are involved in E. histolytica-induced host cell death. For example, intracellular [Ca2+] [6], caspase-3 [7,8], reactive oxygen species (ROS) [9,10,11], protein tyrosine phosphatases (PTP) [12,13], and calpain [14,15] have all been shown to be necessary for E. histolytica-induced host cell death. Therefore, host cell death induced by pathogenic amoebae is closely linked to host tissue inflammation, leading to tissue destruction and the subsequent pathological symptoms of human amoebiasis. However, the detailed signaling mechanisms which underlie amoeba-induced host cell death have not yet been fully elucidated.
Calpain, a calcium-dependent cysteine protease, exists in almost all cells in at least 2 isoforms, µ-calpain and m-calpain [16]. Calpain consists of a large (80 kDa) catalytic subunit and a small (30 kDa) regulatory subunit; the latter is identical in both isoforms [17]. Upon increases in calcium levels, calpains undergo autocleavage to become active proteases, in a similar manner to that of caspases [18]. Calpain activity can be regulated either by autoproteolysis or by an inhibitor protein, calpastatin [19,20]. Accumulating evidence suggests that calpain is involved in several physiological processes such as cell-cycle regulation, transcription factor activation, cell differentiation, and apoptosis [16,21]. Some reports have focused on determining the consequences of excessive calpain activation in various models of cell death [21,22]. For example, calpain activation by either physiological or pathological stimuli has been shown to lead to the degradation of several cytosolic, membrane, and cytoskeleton-associated proteins. Previous studies have also suggested that calpain is responsible for the cleavage of protein tyrosine phosphatases and cytoskeletal-associated proteins during tyrosine dephosphorylation and apoptosis, induced in Jurkat T cells, by E. histolytica [12,13,14]. These results suggest that calpain plays a crucial role in the dismantling of signaling or structural proteins involved in cell survival or integrity during host cell death after exposure to E. histolytica.
Transcription factors play important roles in cell survival and activation. In particular, the NF-κB-directed survival response is associated with increased expression of anti-apoptotic proteins [23]. In contrast, downregulation of NF-κB leads to apoptosis [24]. Signal transducers and activators of transcription (STATs) are members of a family of transcription factors that are activated in response to cytokines and regulate cell proliferation, differentiation, inflammation, the immune response, and fetal development [25]. In particular, STAT3, which is frequently activated in tumor cells, is a valuable target with respect to inhibition of cell proliferation, since its inhibition has been shown to result in colonic carcinoma cell death [26]. A previous report has also indicated that STAT3 and STAT5 are substrates for calpain both in vivo and in vitro [27]. Furthermore, increased calpain activity in neuronal cells has also been shown to induce cell death via degradation of NF-κB (p65) [28]. These results support the hypothesis that E. histolytica-induced activation of calpain can degrade survival factors such as NF-κB or STAT proteins, which may in turn accelerate host cell death. In this study, we used a calpain-specific inhibitor, calpeptin, and siRNA specific for m-calpain to investigate whether calpain plays a role in the E. histolytica-induced degradation of NF-κB, STAT3, or STAT5 in Caco-2 colonic epithelial cells. Our overall aim was to determine the contribution of calpain to cell survival signaling pathways in the context of E. histolytica-induced cell death.
MATERIALS AND METHODS
Reagents
z-VAD-fmk (pan-caspase inhibitor) and calpeptin (calpain inhibitor) were purchased from EMD Biosciences (Darmstadt, Germany). Staurosporin was purchased from Sigma Chemical Company (St. Louis, Missouri, USA). Rabbit polyclonal antibodies against cleaved caspase-3, NF-κB (p65), STAT3, STAT5, and β-actin were purchased from Cell Signaling Technology (Beverly, Massachusetts, USA). Rabbit polyclonal antibodies against IκB-α were purchased from Santa Cruz (Delaware, California, USA). Rabbit polyclonal antibodies against m-calpain and mouse monoclonal antibodies against the calpain regulatory subunit and calpastatin were purchased from Calbiochem (La Jolla, California, USA). Unless stated otherwise, all other reagents were purchased from Sigma-Aldrich (St. Louis, Missouri, USA).
Cultivation of E. histolytica and Caco-2 cells
E. histolytica (HM1:IMSS strain) trophozoites were grown in screw-capped glass tubes containing TYI-S-33 medium at 37℃. After cultivation for 48-72 hr, trophozoites in the logarithmic growth phase were harvested by incubation on ice for 10 min, followed by centrifugation at 200 g at 4℃ for 5 min. Trophozoites were then washed with MEM medium supplemented with 2 g/L NaHCO3, 50 mg/L gentamicin, 1 g/L human serum albumin, and 10% (v/v) heat-inactivated FBS, and subsequently resuspended in culture medium. Caco-2 colonic epithelial cells (American Type Culture Collection, Manassas, Virginia, USA) were maintained in MEM medium containing 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37℃ in a humidified 5% CO2 incubator. Amoebae and Caco-2 cells were always at least 99% viable prior to all experiments, as determined by trypan blue exclusion tests.
Measurements of E. histolytica-induced cell death in Caco-2 cells
Caco-2 cells (5×105 cells) seeded in tissue culture plates and allowed to incubate overnight were subsequently incubated with live E. histolytica trophozoites, at ratios of either 5:1 or 10:1, for 60 min at 37℃ in a CO2 incubator. To assay amoeba-induced DNA fragmentation, Caco-2 cells (4×106 cells) were co-incubated with E. histolytica trophozoites at a ratio of 10:1 for 60 min at 37℃ in a humidified CO2 incubator. After incubation, cells were harvested by centrifugation and subsequently washed with cold PBS. DNA was then extracted using a TaKaRa kit (MK600, Shiga, Japan). DNA samples were then separated by electrophoresis on a 2% agarose gel and subsequently visualized by ethidium bromide staining. LDH release was assessed by determining the amount of LDH in the culture supernatants using the CytoTox 96 Cytotoxicity Assay System (Promega Corporation, Madison, Wisconsin, USA). Culture supernatants were collected after stimulation and subsequently centrifuged at 300 g for 4 min. Supernatants were then incubated with assay buffer and substrate mix at room temperature for 30 min; absorbances at 490 nm were then measured using a 96-well microplate reader. The background absorbance value (corresponding to spontaneous LDH release) was measured in non-stimulated cells and subtracted from each measurement. Maximum LDH release was measured by incubating non-stimulated cells in lysis solution (1% Triton X-100 in PBS) at 37℃ for 45 min.
To determine the role of calpain or caspases in E. histolytica-induced colonic cell death, Caco-2 cells were pretreated with either DMSO (1%, v/v); the pan-caspase inhibitor, Z-VAD-FMK (25, 50, or 100 µM); or the calpain inhibitor, calpeptin (0.25, 0.5, or 1 mM) for 15 min at 37℃. After pre-incubation with inhibitors, cells were washed with cell culture medium and subsequently incubated with E. histolytica. No cytotoxicity was observed for any of the inhibitors at the concentrations tested.
Measurements of caspase-3 activity
To assess caspase-3 cleavage, Caco-2 cells and amoebae were incubated for 15 min at 37℃ in culture medium. Caco-2 cells incubated in medium alone served as a negative control, and treatment with staurosporin (0.5 µM for 6 hr at 37℃) served as a positive control. Western blotting was then performed on proteins from whole cell lysates, using anti-cleaved caspase-3 antibodies.
To directly observe caspase-3 activity in cells being killed by E. histolytica, Caco-2 cells (4×105 cells) were seeded in tissue culture plates 1 day prior to incubation with live E. histolytica trophozoites, at a ratio of 10:1, for 20 min at 37℃ in a CO2 incubator. Following incubation, cells were washed with PBS, fixed with 3% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and immunostained. FITC-conjugated rabbit anti-active caspase-3 monoclonal antibodies (BD Pharmingen, San Diego, California, USA) were used according to the manufacturer's instructions to detect activation of caspase-3 in Caco-2 cells. After a single wash with PBS, caspase-3 activity was measured using a FACScan flow cytometer. Flow cytometric analysis of fluorescence intensity was performed on at least 10,000 cells. As a positive control, cells were incubated with staurosporin.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was obtained from Caco-2 cells using Trizol reagent (Invitrogen Corporation, Carlsbad, California, USA) and reverse-transcribed using a ProSTAR first strand RT-PCR kit (Stratagene, La Jolla, California, USA). PCR was then performed with a specific primer set for m-calpain (m-calpain: 5'-AGAGTCCAGGAGAGGAGAC-3', 5'-ATAAAGTTTTGAGGTGGCAA-3'). Cycling conditions were as follows: 5 min at 95℃, followed by 35 cycles of 30 sec at 95℃, 30 sec at 60℃, and 30 sec at 72℃, with a final amplification of 7 min at 72℃. PCR products were ultimately examined on 2% agarose gels.
siRNA-mediated silencing of m-calpain in Caco-2 cells
The m-calpain siRNA duplexes (Samchully Pharm, Seoul, South Korea) were designed by selecting duplex sequences of the human m-calpain gene (sense: 5'-GUUCAAGACCAUCCAGAAA-3', anti-sense: 5'-UUUCUGGAUGAUCUUGAAC-3'). In mock transfections, all reagents were used except for siRNA. The siRNA cellular transfections were performed using Lipofectamine reagent (Invitrogen) according to the manufacturer's instructions. To optimize siRNA treatment conditions, different concentrations of siRNA (50 or 100 nM) and varying lengths of incubation (24-72 hr) were examined. Throughout all experiments, cell viability was monitored. Cells were viable for the duration of all experiments, as determined by trypan blue exclusion assays (data not shown). At 24, 48, and 72 hr post-transfection, the efficiency of siRNA-mediated m-calpain knockdown was confirmed by both RT-PCR and Western blotting, the latter using m-calpain-specific antibodies and β-actin as a loading control. At 72 hr post-transfection, transfected Caco-2 cells were washed, placed in fresh cell culture medium, and co-incubated with E. histolytica for cell death assays and investigations of signaling molecule statuses.
Western blot analysis
Caco-2 cells (1×106 cells) were lysed in the lysis buffer containing 20 mM Tris-HCl (pH 7.5), 60 mM β-glycerophosphate, 10 mM EDTA, 10 mM MgCl2, 10 mM NaF, 2 mM dithiothreitol, 1 mM Na2VO4, 1 mM 4-amidinophenylmethane sulfonyl fluoride hydrochloride, 1% NP-40, and 5 µg/ml leupeptin on ice for 30 min. After centrifugation at 12,000 g for 5 min, supernatants were harvested, diluted in SDS-PAGE loading buffer, and heated at 100℃ for 5 min. Samples, containing 30 µg total protein, were then resolved on 10% SDS-PAGE gels, transferred to Immobilon P polyvinylidene fluoride membrane (Millipore) by electroblotting, and probed with specific antibodies at 4℃ overnight. Membranes were then incubated with horseradish-peroxidase (HRP)-conjugated anti-rabbit or mouse antibodies at room temperature for 1 hr. Immunoreactivities were then detected using LumiGLO (Cell Signaling).
Statistical analysis
Results are expressed as means±SEM from at least 3 independent experiments. Statistical analyses were conducted using Student's t-tests. Values with P either <0.05 or 0.01 were considered statistically significant.
RESULTS
Incubation with E. histolytica induces cleavage of calpain and calpastatin, but not caspase-3, in Caco-2 cells
We initially examined whether calpain is activated in Caco-2 cells upon co-incubation with live E. histolytica trophozoites. As shown in Fig. 1A, both m-calpain and its small regulatory subunit underwent dose- and time-dependent degradation, beginning at 1 min post-incubation with E. histolytica. The endogenous protein inhibitor, calpastatin, is well known to regulate the activity of calpain [29]. Moreover, calpains improve calpain activity by degrading calpastatin during apoptosis [30]. As shown in Fig. 1B, calpastatin was degraded in a time- and dose-dependent manner in Caco-2 cells upon their co-incubation with E. histolytica. Entamoeba-induced cleavage of calpastatin was inhibited by pretreatment of Caco-2 cells with the calpain inhibitor, calpeptin (Fig. 1C). Next, we examined the activation status of caspase-3 in Caco-2 cells incubated with E. histolytica. As shown in Fig. 1D, Western blot analysis revealed that cleaved forms of caspase-3 are not detected in cell lysates from Entamoeba-treated Caco-2 cells. However, cleaved forms of caspase-3 were clearly found in Caco-2 cells stimulated for 6 hr with 0.5 µM staurosporin. In contrast to Caco-2 cells, Jurkat T cells co-incubated with amoebae did exhibit cleaved forms of caspase-3. Using flow cytometry, we also assessed the intracellular activation of caspase-3 in Caco-2 cells by staining with FITC-conjugated anti-active caspase-3 monoclonal antibodies. When Caco-2 cells were incubated with live trophozoites for 20 min at a 5:1 ratio, the mean fluorescence intensity (MFI) of active caspase-3 was not significantly increased compared with that of cells incubated with medium alone (Fig. 1E). In contrast, intracellular caspase activity was increased 2-fold in Caco-2 cells treated with staurosporin compared with control cells.
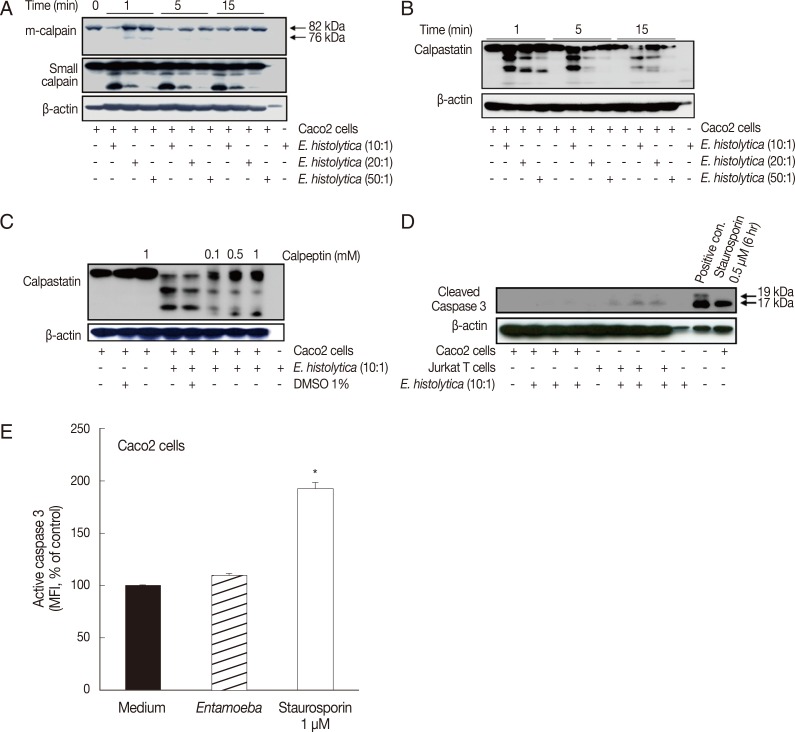
Incubation with E. histolytica induces cleavage of calpain and calpastatin, but not caspase-3, in Caco-2 cells. (A, B) Caco-2 cells (1×106/sample) were incubated for 1-15 min at 37℃, either with or without E. histolytica at ratios of 10:1, 20:1, or 50:1 (Caco-2 cells to E. histolytica). After incubation, proteins in whole cell lysates were subjected to SDS-PAGE and then blotted with anti-m-calpain, anti-calpain small subunit, and anti-calpastatin antibodies (Abs). β-actin was used as a loading control. (C) Caco-2 cells (1×106/sample), pretreated either with or without calpeptin, were incubated for 15 min at 37℃ in either the absence or presence of E. histolytica (1×105/sample) in a CO2 incubator. After incubation, proteins in whole cell lysates were subjected to SDS-PAGE and blotted with anti-calpastatin Abs. β-actin was used as a loading control. (D) Caco-2 cells were incubated with E. histolytica (E. histolytica to Caco-2 ratio, 1:10) at 37℃ for 15 min. Proteins in whole cell lysates were analysed for the presence of activated caspase-3 by immunoblotting with anti-cleaved caspase-3 Abs that recognize the p19 and 17 fragments of caspase-3. Medium alone served as a negative control. Caco-2 cells incubated with staurosporin (1 mM for 6 hr) and Jurkat T cells incubated with amoebae served as positive controls. (E) Caco-2 cells were incubated with E. histolytica (E. histolytica to Caco-2 ratio, 1:10) at 37℃ for 20 min and subsequently stained for activated caspase-3 with FITC-conjugated rabbit anti-active caspase-3 monoclonal antibodies. Activation of caspase-3 was analysed by flow cytometry. Caco-2 cells were incubated in medium alone as a negative control. Staurosporin (1 mM for 6 hr), a known inducer of apoptosis, served as a positive control. Significant differences between groups are indicated as follows: *P<0.005. Figures are representative of three independent experiments, each showing similar results.
E. histolytica induces degradation of NF-κB, STAT3, and STAT5, but not IκB, in Caco-2 cells in a calpain-dependent manner
We next investigated the role of calpain in NF-κB, STAT3, and STAT5 signaling pathways during E. histolytica-induced colonic cell death. First, we performed Western blotting to assess the levels of NF-κB, IκB-α, STAT3, and STAT5 in Caco-2 cells after incubation with E. histolytica. Incubation with E. histolytica induced degradation of NF-κB, STAT3, and STAT5 in a dose-dependent manner, whereas it did not cause any change in the protein levels of IκB-α (the endogenous inhibitor of NF-κB) compared with control cells (Fig. 2A). To determine whether calpain is involved in the mechanism of NF-κB, STAT3, or STAT5 degradation induced by E. histolytica, we next pretreated Caco-2 cells with calpeptin prior to incubation with live trophozoites. As shown in Fig. 2B, pretreatment with calpeptin inhibited E. histolytica-induced degradation of NF-κB, STAT3, and STAT5 in Caco-2 cells. Next, to elucidate the role of m-calpain in the transcription factor degradation induced by the amoeba, Caco-2 cells were transfected with either m-calpain siRNA or scrambled control siRNA. As shown in Fig. 2C, transfection of Caco-2 cells with m-calpain siRNA reduced the mRNA levels of m-calpain mRNA without influencing GAPDH mRNA levels. In contrast, transfection with control siRNA did not affect m-calpain mRNA expression levels. Knockdown of m-calpain was also observed on the protein level upon transfection of m-calpain siRNA, but not with scrambled control siRNA (Fig. 2C). Transfection with m-calpain siRNA also resulted in partial inhibition of E. histolytica-induced degradation of NF-κB, STAT3, and STAT5 in Caco-2 cells (Fig. 2D).
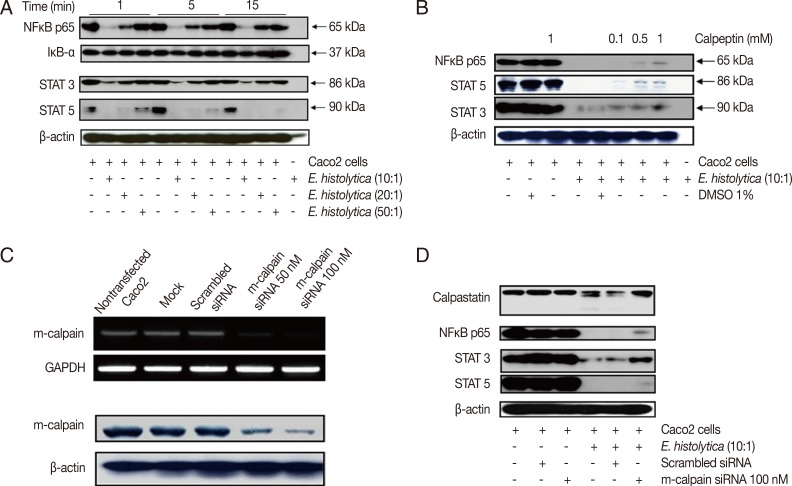
Incubation with E. histolytica induces degradation of NF-κB and STATs, but not IκB-α, in Caco-2 cells in a calpain-dependent manner. (A) Caco-2 cells (1×106/sample) were incubated for 1-15 min at 37℃, either with or without E. histolytica at ratios of 10:1, 20:1, or 50:1 (Caco-2 cells to E. histolytica). After incubation, proteins in whole cell lysates were subjected to SDS-PAGE and subsequently Western blotted with anti-NF-κB (p65), anti-IκB-α, anti-STAT3, and anti-STAT5 Abs. β-actin was used as a loading control. (B) Caco-2 cells (1×106/sample), pretreated either with or without calpeptin, were incubated for 15 min at 37℃ in either the absence or presence of E. histolytica (1×105/sample) in a CO2 incubator. After incubation, proteins in whole cell lysa tes were subjected to SDS-PAGE and Western blotted with anti-NF-κB (p65), anti-STAT3, and anti-STAT5 Abs. β-actin was used as a loading control. (C) Downregulation of both mRNA expression levels and protein levels upon transfection of m-calpain siRNA into Caco-2 cells. At 72 hr post-transfection, cDNA and proteins in whole cell lysates from Caco-2 cells transfected with either vehicle alone (mock), scrambled siRNA (negative control), m-calpain siRNA, or nothing (nontransfected) were subjected to RT-PCR and immunoblotting with anti-m-calpain Abs, respectively. β-actin was used as a loading control. (D) At 72 hr post-transfection, proteins in whole cell lysates from Caco-2 cells transfected either with nothing (nontransfected), scrambled siRNA, or m-calpain siRNA, and co-incubated either with or without Entamoeba histolytica, were subjected to SDS-PAGE and subsequently probed with anti-calpastatin, anti-NF-κB (p65), anti-STAT3, and anti-STAT5 Abs. β-actin was used as a loading control. Figures are representative of three independent experiments, each showing similar results.
m-calpain is required for E. histolytica-induced cell death in Caco-2 cells
Finally, we examined the role of calpain in E. histolytica-induced cell death in Caco-2 cells. As shown in Fig. 3A, incubation of Caco-2 cells with calpeptin for 15 min prior to exposure to E. histolytica effectively inhibited E. histolytica-induced release of LDH from Caco-2 cells, compared with cells pretreated with 1% DMSO. However, this pro-death effect of amoebae on LDH release in Caco-2 cells was not prevented by pretreatment with 100 µM z-VAD-fmk (Fig. 3B). We further examined the involvement of calpain in E. histolytica-induced cell death in Caco-2 cells. As shown in Fig. 3C and D, E. histolytica-induced DNA fragmentation in Caco-2 cells was significantly diminished either by treatment with the calpain inhibitor, calpeptin, or by transfection with m-calpain siRNA, compared with controls.
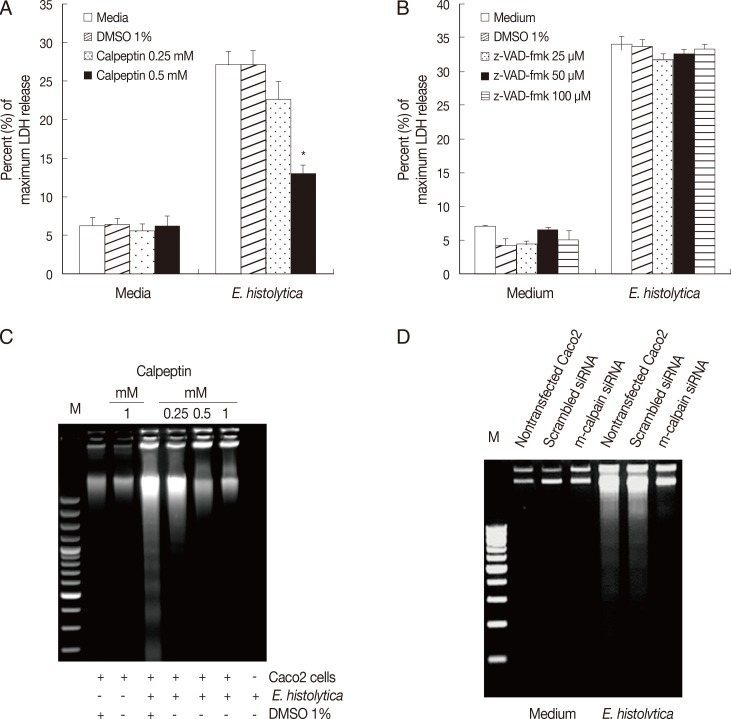
m-calpain is required for E. histolytica-induced cell death in Caco-2 cells. (A) Caco-2 cells (3×105/sample) pretreated with either calpeptin (0.25-0.5 mM) or DMSO (1%) were incubated for 60 min with E. histolytica at a ratio of 5:1. Cell death was measured by the LDH release assay. Significant differences between groups are indicated as follows: *P<0.005. (B) Caco-2 cells (3×105/sample) were incubatedfor 60 min with E. histolytica, at a ratio of 5:1, in either the absence or presence of z-VAD-fmk (25-100 mM). Cell death was measuredby the LDH release assay. Data are presented as means±SEM from three independent experiments. (C) Caco-2 cells (2.5×106/sample) pretreated either with or without calpeptin (0.25-1 mM), or 1% DMSO (v/v) as a control, for 15 min at 37℃ were then incubated for 60 min at 37℃ in a CO2 incubator in either the absence or presence of E. histolytica (2.5×105/sample). DNA fragmentation was subsequentlyanalysed by electrophoresis on 2% agarose gels. (D) At 72 hr post-transfection, Caco-2 cells were transfected with either scrambled siRNA (negative control) or m-calpain siRNA and subsequently co-incubated with E. histolytica prior to analysis of cell death by DNA fragmentation. Nontransfected Caco-2 cells served as an additional control.
DISCUSSION
Although the signaling pathways involved in Entamoeba-induced cell death have been studied by many investigators in multiple cell lines, the precise mechanism by which Entamoeba induces cell death in Caco-2 colonic epithelial cells has not yet been completely elucidated. In this study, we demonstrated that calpain-mediated degradation of multiple transcription factors is involved for E. histolytica-induced cell death in Caco-2 colonic epithelial cells.
Calpain is a critical mediator of cell death and is triggered by calcium signals; the ability of calpain to cleave a growing number of substrates also suggests a potentially important role for this enzyme in the regulation of cell death. Based on previous studies reporting a critical role for calpain in E. histolytica-induced cell death of Jurkat T cells [8], we postulated that calpain regulates Entamoeba-induced degradation of transcription factors in Caco-2 cells. Our results demonstrated that incubation with live trophozoites induced activation of calpain and degradation of NF-κB, STAT3, and STAT5 in Caco-2 cells, whereas it did not trigger any change in the protein levels of IκB-α. Also, E. histolytica-induced degradation of transcription factors was inhibited by calpain inactivation with calpeptin. Furthermore, both calpeptin treatment and siRNA-mediated knockdown of m-calpain reduced E. histolytica-induced cell death in Caco-2 cells. Therefore, our results suggest that calpain facilitates cell death via degradation of transcription factors in Caco-2 cells in contact with E. histolytica.
Unlike caspases, which require specific amino acid sequences at their cleavage sites, calpains show no apparent sequence preferences. Calpain has a large number of substrates, including signaling molecules, membrane proteins, intracellular enzymes, and structural proteins [16,31,32,33,34,35]. Calpain may play a role in apoptosis by cleaving cytoskeletal proteins such as fodrin and actin, in addition to cleaving pro-apoptotic proteins [36,37]. Interestingly, Gao and Dou [38] reported that calpain can cleave Bax, thereby generating a Bax/p18 fragment which mediates cytochrome c release and initiates execution of apoptosis.
The transcription factor, NF-κB, plays an important role in cell survival. In many cancers, upregulation of NF-κB exerts anti-apoptotic effects which lead to tumor cell survival, transformation, and resistance to radiation and drug treatments [39]. In contrast, downregulation of NF-κB leads to apoptosis [24]. Upregulation of NF-κB has been reported to result in overexpression of survival factors which suppress caspase activation and inhibit apoptosis [40]. On the other hand, several studies have shown that caspase-dependent degradation of NF-κB can reduce inflammatory responses in macrophages [41] and induce cell death in endothelial cells [42] and renal cells [43]. Our investigation demonstrated for the first time that contact with E. histolytica decreased protein levels of NF-κB, via a calpain-dependent manner, in Caco-2 cells (Fig. 2B). Interestingly, incubation with amoebae did not impair the endogenous inhibitor of NF-κB, IκB-a, in Caco-2 cells (Fig. 2A). These results suggest that E. histolytica actively induces cell death in Caco-2 cells.
Signal transducers and activators of transcription (STATs) comprise a family of transcription factors whose members are activated in response to cytokines and regulate cell proliferation, differentiation, inflammation, the immune response, apoptosis, and fetal development [25]. A previous report indicated that STAT3 is a substrate for calpain both in vivo and in vitro, implying that calpain-mediated cleavage is a common feature of STAT3 and STAT5 [27]. The role of STAT3 in malignant cell growth is mediated in part by the upregulation of genes involved in cell survival and proliferation, including Bcl-xl, Bcl-2, c-Myc, cyclin D1, survivin, Mcl-1, vascular endothelial growth factor, IL-10, and transforming growth factor [44,45,46,47,48]. In addition to STAT3 and STAT5, regulation by degradation has also been reported for STAT6 [49]. Furthermore, Datta et al. [50] reported that calpain blocks IL-5 expression by degrading STAT6 in PARP-1 deficient splenocytes. The constitutive activation of STATs observed in many tumours and tumour cell lines suggests that it may be a good target for the induction of cell death.
To determine whether calpain plays a role in E. histolytica-induced cleavage of STAT3 and STAT5, we pretreated Caco-2 cells with a calpain inhibitor, calpeptin. Our data indicated that pretreatment of Caco-2 cells with calpeptin blocked E. histolytica-induced cleavage of these STATs, in a dose-dependent manner. Given that calpain has been implicated in many cellular processes, including cell proliferation, apoptosis, and differentiation [51], cleavage of STAT5 by activated calpain may also inhibit STAT5-mediated signaling in additional situations. For example, strong T cell receptor (TCR) signaling, which is likely to increase intracellular free calcium and thereby induce more calpain activation, is well known to drive Th1 polarization [52]. Since STAT5a has been suggested to play a critical role in Th2 cell differentiation [53,54], it is possible that TCR-mediated calpain activation induces STAT5a cleavage, thereby impairing Th2 cell differentiation. Our findings suggest that calpain is capable of downregulating multiple survival factors, thereby inhibiting cell survival signals and consequently promoting Caco-2 cell death induced by E. histolytica. Although it is uncertain how degradation of these transcription factors would contribute to the rapid cell death observed in this study, degradation of these transcription factors may be a major factor in modulating the host response during in vivo infection. Accordingly, we attempt to perform this work to include an analysis of the transcriptional targets of NFKB/STAT3/STAT5 subsequently.
Here, we have observed that the co-incubation with Entamoeba had no effect on caspase-3 activation. In addition, in this study, Bax and Bid were not cleaved in Caco-2 cells following incubation with E. histolytica (data not shown). These results support that the caspase-9/-3 apoptotic pathway is not involved in Entamoeba-induced Caco2 cell death model. Previously Kim et al. [11] reported that caspase-3 works independently in the process of HT29 cell death induced by E. histolytica. Also, when we used Caco-2 colon epithelial cells, a previous report has shown that NADPH oxidase inhibitor or NOX1 siRNA reduced E. histolytica-induced cell death [10], suggesting that calpain as well as ROS may participate in the process of Caco-2 cell death. Although it is apparent that calpain can regulate activation of caspase-3 in Jurkat T cells [8], pretreatment with calpain inhibitor did not work to inhibit cell death induced by E. histolytica. Caspase-3-dependent apoptotic cell death in Jurkat T cells by E. histolytica might contribute to little inflammatory responses leading to silence host immune responses and minimizing tissue damage, which may be beneficial to maintain chronic infection during extraintestinal amoebiasis. Whereas E. histolytica-induced colon cell death is directly associated to provocation of serious tissue inflammation and extensive tissue damage of the amoeba-infected colon lesions. These results suggest that signaling molecules involved in cell death induced by E. histolytica may vary in different cells.
In conclusion, this study has shown for the first time that calpain-mediated degradation of transcription factors is associated with E. histolytica-induced colonic epithelial cell death. Further studies on the detailed regulatory role of calpain in this process will assuredly provide important information regarding amoeba-induced inflammatory responses in human amoebiasis, and contribute to the development of therapeutic strategies.
ACKNOWLEDGMENTS
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MEST) (no. 2012RIA2A2A02015054).
Notes
We have no conflict of interest related to this work.