Purification and biochemical characterization of two novel antigens from Leishmania major promastigotes
Article information
Abstract
The identification and characterization of antigens that elicit human T cell responses is an important step toward understanding of Leishmania major infection and ultimately in the development of a vaccine. Micropreparative SDS-PAGE followed by electrotransfer to a PVDF membrane and elution of proteins from the PVDF, was used to separate 2 novel proteins from L. major promastigotes, which can induce antibodies of the IgG2a isotype in mice and also are recognized by antisera of recovered human cutaneous leishmaniasis subjects. Fractionation of the crude extract of L. major revealed that all detectable proteins of interest were present within the soluble Leishmania antigens (SLA). Quantitation of these proteins showed that their expression in promastigotes is relatively very low. Considering the molecular weight, immunoreactivity, chromatographic and electrophoretic behavior in reducing and non-reducing conditions, these proteins are probably 2 isoforms of a single protein. A digest of these proteins was resolved on Tricine-SDS-PAGE and immunoreactive fragments were identified by human sera. Two immunoreactive fragments (36.4 and 34.8 kDa) were only generated by endoproteinase Glu-C treatment. These immunoreactive fragments or their parent molecules may be ideal candidates for incorporation in a cocktail vaccine against cutaneous leishmaniasis.
Infection of humans by protozoa of the genus Leishmania results in a clinical spectrum of cutaneous, visceral, and mucosal diseases, which together constitute an important world-wide public health problem. Experimental murine models with high, low, and intermediate levels of genetically-based susceptibility to Leishmania infection have reproduced the entire spectrum of clinical manifestations of the human disease with the exception of the mucosal form (Gumy et al., 2004). For example, BALB/c mice infected in the footpad with Leishmania major (the causative agent of cutaneous leishmaniasis) develop large ulcerating lesions, with the eventual necrosis of the foot, visceralization of the parasite, and death of the animal, whereas resistant murine strains, such as C57BL/6, control parasite growth, resolve their lesions, and become immune to reinfection. In addition, the mouse model of L. major is distinctive in that certain strains of mice (C57BL/6 and C3H) develop protective T helper-1 (Th1) responses after infection, whereas other strains (BALB/c) develop T helper-2 (Th2) responses and succumb to infection (Papadopoulou et al., 1998; Sacks and Anderson, 2004). However, Th1 cells are also found in BALB/c mice with a predominant Th2 response.
Regardless of clinical presentation, recovery and resistance to reinfection are dependent on T helper cells (Heinzel et al., 1991; Skeiky et al., 1998). Protection depends on the activation of Th1 cells producing IFN-γ, but not IL-4, whereas induction of Th2-like T cells secreting IL-4 results in the inability to control disease or disease exacerbation. The outcome of Leishmania infection thus depends on the early cytokine profile. Therefore, parasite molecules that influence early cytokine production may be key determinants of resistance or susceptibility.
Qualitative studies on Leishmania antigens on immunoblots have demonstrated preferential recognition of Leishmania infantum, Leishmania donovani, and L. major antigens by a particular subclass of antibodies (Shimizu et al., 2003; Iborra et al., 2004; Lange et al., 2004; Tewary et al., 2004). The isotype profile of antibody responses depends on the cytokines produced by antigen specific T-cells. Production of IgG2a is dependent on IFN-γ whereas IL-4 is important for the generation of high levels of IgG1 (Snapper and Paul, 1987; Papadopoulou et al., 1998). The relative production of these isotypes can thus be used as a marker for the induction of Th1-like and Th2-like immune responses, respectively.
The identification and characterization of L. major antigens that elicit T cell responses is an important step toward understanding the immunology of cutaneous leishmaniasis and in the development of a vaccine. Previously we have shown that 2 novel antigens from L. major promastigotes in the late log-phase of growth are capable to induce antibodies of the IgG2a isotype in both resistant and susceptible mice (Mohammadi et al., 2006). The goal of the current study is to localize and further characterize these novel antigens. Western blotting of proteins from SDS-PAGE to a PVDF membrane, and protein elution from the membrane was used for co-purification of these parasite antigens. Gel electrophoresis and high-performance liquid chromatography in reducing and non-reducing condition, protein digestion with different endoproteinases and immunoblotting were used for partial characterization of these novel antigens.
A clone of L. major (MRHO/IR/75/ER strain) was used for this study. Parasites were grown at 27℃ in RPMI 1640 medium supplemented with 10% fetal bovine sera (FBS), 2 mM glutamine, 100 U/ml penicillin G-potassium, 100 µg/ml streptomycin sulfate and 25 mM HEPES. To prepare soluble Leishmania antigen (SLA), promastigotes at late logarithmic phase were harvested from culture, washed 4 times in cold PBS (pH 7.7) and resuspended at 1.2 × 109 parasites/ml in PBS containing 0.1 mM EDTA, 0.5 mM DTT and 1 mM p-APMSF. The parasites were then subjected to sonication at 4℃with 2 times 30-sec blasts. The parasite suspension was then centrifuged at 27,000 g for 30 min at 4℃ The supernatant was collected and re-centrifuged at 100,000 g for 1 hr. The supernatant was then collected and stored as SLA at -20℃ until use. The residual pellet was washed in 0.25 M sucrose containing 0.15 M NaCl and 5 mM Tris-HCl, pH 8. The cell membranes were then pelleted by centrifugation at 100,000 g for 1 hr. Membrane content was resuspended to a protein concentration of 1-2 mg/ml in a solution containing 1% (w/v) of SDS, 10% sucrose and 0.1 M Tris-HCl, pH 8.5. The mixture was kept at room temperature for about 15 min and then centrifuged at 100,000 g for 1 hr. The clear supernatant (membrane fraction) was collected and then diluted with an equal volume of 2x Laemmli sample buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, and 0.004% bromophenol blue). Protein concentration of SLA and membrane fractions was determined by a modified Lowry assay (Sandermann and Strominger, 1972).
Reducing and non-reducing SDS-PAGE were performed on a slab gel apparatus utilizing the discontinuous system (Laemmli, 1970). The separated protein bands were stained using either the Coomassie blue or silver staining techniques (Helmut, 1987). For blotting, a semi-dry assembly was prepared according to Kyhse-Andersen (1984). Electrotransfer was performed for 2 hr at 1.5 mA/cm2. Following blotting, 5mm strips of PVDF with blotted proteins were cut out and blocked for 1 hr at room temperature in washing buffer (10 mM Tris-HCl, 0.15 M NaCl, 0.3% Tween-20, pH 7.5) containing 3% BSA and then incubated overnight at 4℃ with sera from recovered cutaneous leishmaniasis human subjects (1: 20 dilution). The strips were then washed 3 times with washing buffer for 10 min and incubated for 2 hr at room temperature in horseradish peroxidase-conjugated anti-human IgG (Sigma, St. Louis, Missouri, USA) diluted in washing buffer (1: 1,000 dilution). Unbound secondary antibodies were removed by washing 3 times for 10 min. The membrane was developed by incubating the strips few minutes with substrate solution (50 mM Tris-HCl, pH 7.5, 0.03% diaminobenzidine, 0.08% H2O2).
For protein purification, the antigenic bands of interest were excised from PVDF membranes and placed in a tube containing 0.2-0.5 ml of a detergent solution (2% SDS, 1% Triton X-100, 0.1% DTT in 50 mM Tris-HCl, pH 9.0) per cm2 of the membrane. Elution procedure was performed according to Simpson et al. (1989). The eluent was filtered on a YM-10 membrane filter (Millipore, Bedford, Massachusetts, USA) and then precipitated by cold acetone. The pellet was resuspended in a small volume of 20 mM Tris-HCl, pH 7.5 and the protein concentration was determined by the Bradford assay.
RP-HPLC analyses of the eluent were conducted on a C4-column (4.6 × 250 mm). Solutions A and B were 0.1% trifluoroacetic acid (TFA) in ddH2O and 0.1% TFA in acetonitrile, respectively. The protein preparation was loaded onto the C4-column and bound molecules were eluted by a linear 0-100% gradient of B solution at a flow rate of 0.8 ml/min. The effluent was monitored continuously at 215 nm.
For digestion experiments, the protein preparation was dissolved at approximately 50 µg/ml of sample buffer (0.125 M Tris-HCl, pH 6.8, 0.1% SDS, 10% glycerol, and 0.001% bromophenol blue). The samples were then heated to 100℃ for 2 min. Proteolytic digestions were carried out at 25℃ for 16-24 hr by addition of 2.5-5 µg/ml of any of 4 proteases (Roche, Mannheim, Germany); endoproteinase Lys-C, endoproteinase Asp-N, endoproteinase Glu-C, and trypsin. Following addition of 2-mercaptoethanol and SDS to a final concentration of 10% and 2%, respectively, proteolysis was stopped by boiling the samples for 2 min. The protein lysates were analyzed on Tricine-SDS-PAGE according to Schägger and von Jagow (1987). Silver staining of gel was performed according to Oakley et al. (1980). Western blotting of the Tricine-SDS-PAGE was performed according to Kyhse-Andersen (1984).
Isoelectric focusing was performed according to Ames and Nikaido (1976). Samples were mixed with an equal volume of lysis buffer (9.4 M urea, 2% NP-40, 2% Ampholine pH range 3-10, 5% 2-mercaptoethanol) and loaded onto wells. Electrophoresis was conducted at 400 volts for 12 hr, then 600 volts for 1 hr. For staining, gel firstly was placed in 10% TCA for 2 hr and then silver staining was performed according to Oakley et al. (1980).
Two antigenic bands, 140 and 152 kDa in size, were identified in crude extracts of L. major (Fig. 1A) by immunoblotting using sera from recovered cutaneous leishmaniasis subjects. Fractionation of the crude extract of L. major and immunoblot analysis was performed in order to determine the location of these 2 novel antigens. It was revealed that all detectable 152 and 140 kDa proteins were present within the SLA fraction (Fig. 1B) but were absent from the cellular membranes (Fig. 1C). A total of 2.1 × 1010 parasites yielded approximately 160 µg of these 2 proteins.
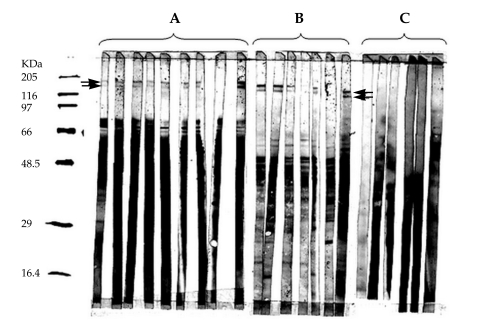
Detection of the 152 and 140 kDa proteins in soluble Leishmania antigens (SLA). Immunoblot analysis of L. major promastigotes lysate (A), SLA (B) and membrane fraction (C) of L. major, which were resolved on a 10% homogeneous SDS-PAGE, transferred onto a PVDF membrane, probed with sera from recovered cutaneous leishmaniasis subjects and then detected with horseradish peroxidase-conjugated anti-human IgG and stained calorimetrically. Lane 1 is molecular weight marker and the numbers represent molecular weight standards. Arrows indicate the location of 152 and 140 kDa antigenic bands.
On SDS-PAGE under reducing conditions, the purified sample showed a diffuse band with an apparent molecular weight of 65 kDa (Fig. 2). Considering the Helmut silver staining technique, which can detect impurities as low as l ng, it was concluded that the level of trace impurities, if any, in 2 µg of co-eluted proteins were less than 1 ng. IEF analysis of the co-eluted proteins also indicated a single band of a pI about 4.2. Analysis of the protein preparation by an analytical RP-HPLC column under reducing and non-reducing conditions gave 2 overlapping peaks (Fig. 3). In the reducing condition, the retention time of the main peak (peak I) changed from 21.89 to 10.78 min.
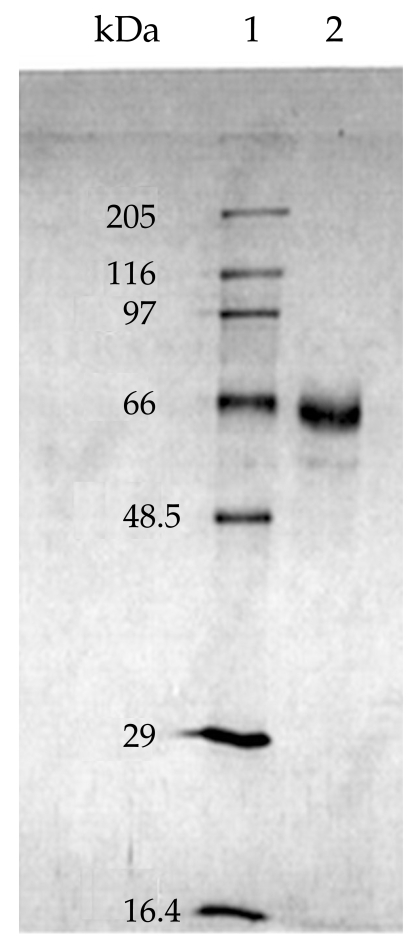
SDS-PAGE analysis of the co-purified 152 and 140 kDa proteins. Lane 1, molecular weight marker and Lane 2, mixture of the purified novel antigens under reducing condition.
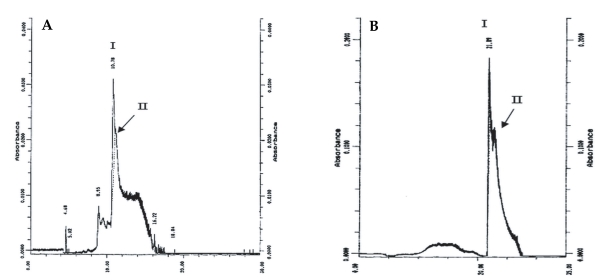
RP-HPLC analysis of the co-purified proteins in reducing (A) and non-reducing (B) conditions. Mixture of the 152 and 140 kDa proteins was loaded onto a C4 column. Chromatographic conditions were flow rate 0.8 ml/min and 0-100% linear gradient of acetonitrile. The retention time of the main peak (peak I) changed from 21.89 to 10.78 min in the presence of a reducing agent.
Analysis of the protein preparation by immunoblotting with human sera following SDS-PAGE in a reducing condition showed 65.43 and 62.96 kDa antigenic bands, whereas in a non-reducing environment the 140- and 152-kDa doublets were present.
After incubating the co-eluted proteins for 16 hr at 25℃ in the presence of 2.5 µg/ml of each of the above mentioned proteinases, the generated peptides were analyzed by Tricine-SDS-PAGE. The results of gel analysis showed that the co-eluted antigens were partially proteolyzed by each of these enzymes after 16 hr (Fig. 4). Because of incomplete digestion of original doublets by Glu-C endoproteinase and trypsin after 16 hr, the concentrations of these enzymes were increased to 5 µg/ml and digestion time was increased to 24 hr. Immunoreactive fragments (36.4 and 34.8 kDa) were only generated by endoproteinase Glu-C treatment (Fig. 5).
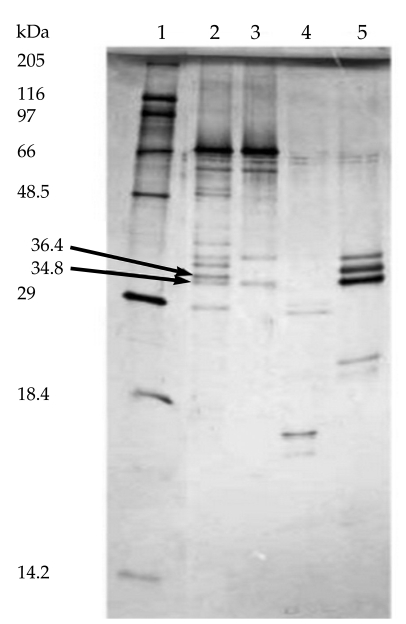
Proteolytic degradation of the co-purified proteins with 4 different endoproteinases. Lane 1, molecular weight marker and Lanes 2 to 5, treated novel antigens with Glu-C endoproteinase, Trypsin, Lys-C endoproteinase and ASP-N endoproteinase, respectively.

Immunoblot analysis of the digested proteins. Treated antigens with Glu-C endoproteinase (Lanes 1 and 2), trypsin (Lanes 3 and 4), Asp-N endoproteinase (Lanes 5 and 6) and Lys-C endoproteinase (Lanes 7 and 8) have been shown. Sera from recovered cutaneous leishmaniasis subjects were used as primary antibodies. Arrows indicate the location of 36.4 and 34.8 kDa antigenic fragments.
Previously, we showed that 152 and 140 kDa proteins from the L. major promastigotes were capable to induce antibodies of the IgG2a isotype in both susceptible and resistant mice (Mohammadi et al., 2006). In the present study, we made attempts to purify these 2 recently detected novel antigens from L. major promastigotes by a combination of SDS-PAGE and western blotting. A crude extract of L. major promastigotes was fractionated into SLA and membrane fractions. The novel proteins, 152 and 140 kDa, were detected in SLA (Fig. 1B). Because of very low level of cellular expression of these proteins (0.48% of total SLA) in L. major, utilization of techniques, such as chromatography, which needs large amounts of samples, would be challenging and uneconomical. In addition, recovery during chromatographic processes is often inadequate. Electrophoresis on the SDS-PAGE, combining with protein elution from PVDF membranes can be an efficient and cost-effective way to purify denatured proteins for immunological analysis and microsequencing. For protein identification purpose, the 2 antigenic proteins were co-purified from a PVDF membrane, since both proteins migrated closely to each other on gels, and their expression levels were relatively low in the parasites. After purification, re-electrophoresis on SDS-PAGE in reducing condition and immunoblotting, it was revealed that molecular weights of the purified proteins are roughly half (62-65 kDa) of their original size. In contrast, SDS-PAGE under non-reducing condition restored the size of the original doublets. This suggests that the 152 and 140 kDa proteins are probably dimers of identical subunits hold in conformations by disulfide-bridges that become dissociated by reducing agents, such as DTT in denaturing environments of SDS and Triton X-100. This observation was corroborated by RP-HPLC in C4 column that was also used to confirm the protein purity.
The results of RP-HPLC analyses under both reducing and non-reducing conditions showed that the retention times of 2 overlapping chromatographic peaks decrease 2-fold at reducing conditions, reminiscent of the electrophoretic results. Considering the molecular weight, immunoreactivity, chromatographic and electrophoretic behaviors in different conditions, it is probable that these proteins are 2 isoforms of a single protein, although the N-terminal sequences of 140 and 152 kDa proteins need be completed to establish true identity of these proteins.
In an attempt to identify the antigenic domains of purified proteins recognized by human sera, partial proteolytic digestion of purified proteins was performed by 4 sequencing grade endoproteinases, and the cleavage products were analyzed by immunoblotting. Immunoreactive fragments (36.4 and 34.9 kDa) were only generated by endoproteinase Glu-C (V8-protease) treatment. Further fragmentation of these immunoreactive bands by other proteases resulted in band disappearing. Smaller peptides were not recognized by human sera.
In summary, biochemical and immunological procedures were utilized in the present study for micropreparative co-purification and partial characterization of 2 novel L. major proteins from SLA fraction. These antigens were previously found to induce an IgG2a response in mice (Mohammadi et al., 2006) and may elicit Th1 cell responses in humans. Because of relatively low expression of these proteins in L. major, cloning and expression of full length proteins or immunoreactive fragments should be used for a detailed analysis of their role in eliciting potentially protective CD4 Th1 cell responses and should ultimately contribute to the development of a defined L. major vaccine.
ACKNOWLEDGMENTS
This work was financially supported by grants from the Research Council of University of Tehran, Iran.