Complete Mitochondrial Genome of a Tongue Worm Armillifer agkistrodontis
Article information
Abstract
Armillifer agkistrodontis (Ichthyostraca: Pantastomida) is a parasitic pathogen, only reported in China, which can cause a zoonotic disease, pentastomiasis. A complete mitochondrial (mt) genome was 16,521 bp comprising 13 protein-coding genes (PCGs), 22 tRNA genes, 2 rRNA genes, and 1 non-coding region (NCR). A phylogenetic tree drawn with the concatenated amino acid sequences of the 6 conserved PCGs (atp6, cox1-3, and nad2) showed that A. agkistrodontis and Armillifer armillatus constituted a clade Pentastomida which was a sister group of the Branchiura. The complete mt genome sequence of A. agkistrodontis provides important genetic markers for both phylogenetic and epidemiological studies of pentastomids.
Human pentastomiasis is a zoonotic parasitic disease, which is caused by infection with the nymphal stage of the pentastomid. Although more than 10 pathogenic species associated with human disease have so far been identified, Linguatula serrata and Armillifer armillatus are implicated in the majority of cases [1,2]. Two of these pathogenic species, Armillifer moniliformis and Armillifer agkistrodontis, are found exclusively in China, where the pit viper snake (Deinagkistrodon acutus) is the most common final host for A. agkistrodontis [3]. Infections are acquired by the ingestion of snake meat, blood, and bile contaminated with mature eggs containing the primary larva. Recently, there has been an increase in infection cases of A. moniliformis and A. agkistrodontis raising the profile of this previously neglected disease [2,4,5].
The nymphs that cause human visceral pentastomiasis are mainly located in or on the liver, the intestinal wall, and mesentery organs, as a calcified cyst. Since the characteristics of the nymph are not completely developed, it is necessary to combine both morphological and molecular approaches to facilitate the disease diagnosis and species identification. The sequence of the mitochondrial (mt) genome has been widely used as a specific molecular marker for species identification as it contains more information than single genes or fragments [6].
In this work, we have released the complete sequence for the mt genome of A. agkistrodontis, annotated for gene order and codon usage for protein-coding genes (PCGs). Furthermore, we have attempted to use of the mt PCGs data for phylogenetic analysis to define the relationship with other arthropods, especially Mandibulata and Pancrustacea. We have provided evidence, based on the analysis of this mt genome, supporting the placement of Pentastomida as a sister group to Branchiura within the Pancrustacea clade. This result suggested that the best strategy for utilizing the mt genome for phylogenetic analysis should be the 6 conserved PCGs (atp6, cox1-3, and nad2).
Adult individuals of A. agkistrodontis were collected from the visceral organ and air sac of captive D. acutus purchased from the market in Taizhou city, Zhejiang province, P. R. China. These pentastomids were washed with saline buffer (0.7% NaCl) before being fixed in 70% (v/v) ethanol at 4°C. Species and sexual identification of pentastomids was defined by morphological characteristics [7], after which the tegument was isolated from individual females by excision under a stereomicroscope. Total genomic DNA was extracted from this tegument tissue using a DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and stored at −20°C until use. The identity of A. agkistrodontis specimens was also confirmed by sequencing a gene fragment, coding for 18S rRNA, amplified by PCR from total DNA [3].
The complete mt genomic sequence of A. agkistrodontis was amplified, as described previously, in 6 overlapping fragments, using 6 pairs of oligonucleotide primers designed based on the conserved regions from published complete mtDNA sequences of A. armillatus (no. NC_005934.1) (Table 1) [8,9]. These amplicons were sequenced in both directions by primer walking using an ABI 3730 DNA analyzer (Genewiz, Inc., Suzhou, China).

Primer sequences used to amplify PCR fragments of Armillifer agkistrodontis
The complete mt genome sequence of A. agkistrodontis was assembled manually from individual sequence runs and then aligned against the mt genome sequences of A. armillatus by the program MAFFT 7.122. Thirteen PCGs were identified and translated into amino acid sequences using MEGA 6.0 with the invertebrate mitochondrial code option. The tRNA genes were identified using online services ARWEN (http://130.235.46.10/ARWEN/) and tRNAscan-SE (http://selab.janelia.org/tRNAscan-SE/). tRNA genes that could not be determined by either program, were identified by comparison with the annotations of for the A. armillatus mt genome.
To investigate the sequence similarity, 12 concatenated amino acid sequences translated from each of 12 PCGs, except atp8, from the mt genomes were aligned with those of 21 published mt genomes from selected Arthropoda species: Argulus americanus, Ammothea carolinensis, A. armillatus, Anopheles quadrimaculatus, Abacion magnum, Blattella bisignata, Calanus hyperboreus, Colossendeis megalonyx, Daphnia magna, Euphausia pacifica, Hutchinsoniella macracantha, Ixodes ricinus, Lepas australis, Nobia grandis, Rhipicephalus microplus, Scutigerella causeyae, Strigamia maritima, Sericinus montela, Trichophthalma punctate, Opisthopatus cinctipes, and Metaperipatus inae.
Alignments of both nucleotide and amino acid sequences were performed using Muscle 3.8.31 [10]. The amino acid sequences were extracted from 22 selected mitochondrial genomes (including our data), and 12 of the PCGs, except for atp8, were aligned. The nucleotide sequence from the genes most conserved (atp6, cox1-3, and nad2) in arthropod species [11] were aligned, using the amino acid alignment as a guide, with online software TranslatorX [12]. All the ambiguous gaps were excluded using GBlock [13], resulting in 3870 bases in the final nucleotide alignment.
Amino acid sequence sets were compared using a Bayesian phylogenetic analysis with a mtREV replacement model where the gamma distribution and a fraction of sites were constrained to be invariable. In order to minimize long-branch bias, a Neutral Transitions Excluded method (NTE) was used for this phylogenetic analysis [14]. All Bayesian analyses were generated by MrBayes 3.2 with 5 independent Markov chains run for 2,000,000 generations, a sampling frequency of 1,000 and a burn-in of 250 trees [15].
The complete mt genome of A. agkistrodontis was 16,521 bp in size (no. KX686568), slightly smaller than that of A. armillatus (16,747 bp), with a difference in length mainly due to the non-coding region (NCR). The gene content was typical, including 13 PCGs (atp6, atp8, cox1-3, nad1-6, cytb, and nad4L), 22 tRNA genes, 2 rRNA genes (rrnS and rrnL), and 1 NCR (Fig. 1; Table S1). The gene order and transcriptional orientation were identical to that of A. armillatus, which has been hypothesized to be the ancestral arrangement for the mt genome of insects [8]. The overlapping nucleotides between mt genes in A. agkistrodontis ranged from 1 to 22 bp, the longest overlap located between cytb and nad1 (Table S1). The nucleotide content was 34.20%, 27.33%, 31.01%, and 7.46%, for A, C, T, and G, respectively, with the overall A+T content being 65.21%, ranging from 61.44% (cytb and nad1) to 72.97% (atp8) across the 13 PCGs, 2 rRNA genes, and NCR.
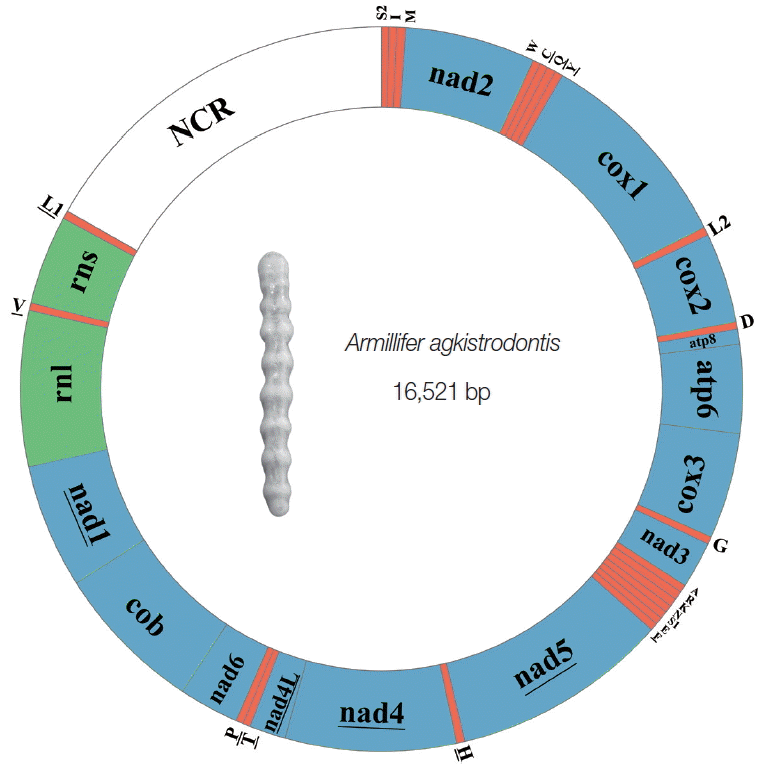
The mitochondrial genome of Armillifer agkistrodontis. The underlined genes are transcribed anticlockwise, while those without underlining are transcribed clockwise. All the tRNA genes are indicated with the 1-letter code of their corresponding amino acids. There are 2 tRNA genes for leucine: L1 for codons CUN and L2 for UUR; and 2 tRNA genes for serine: S1 for codons AGN and S2 for UCN. “NCR” refers to the non-coding region.
The 13 PCGs, 10,652 bp in length (including the terminator codons) accounted, for 64.46% of the entire mt genome. These PCGs had a consensus start codon of ATN (1 with ATT, 2 with ATC, 4 with ATA, and 5 with ATG) with only 1 being CTG (cox1). Conventional stop codons of TAA and TAG were found in 8 of 13 PCGs; however, 5 PCGs (cox1, cox2, cox3, nad4, and nad5) had incomplete termination codons (T) (Table S1). The codon usage pattern of the 13 PCGs is shown in Table S1, with A+T rich codons most frequently used; ATT (Ile: 5.24%), TTA (Leu: 4.90%), and ATA (Met: 4.68%). The least used codons were CGG (Arg: 0.11%), ACG (Thr: 0.11%), and GCG (Ala: 0.17%).
The 22 tRNA genes typically found in metazoan mt genomes were also identified in A. agkistrodontis, ranging in length from 53 bp to 65 bp (Table S1), with 16 of them determined by ARWEN and tRNAscan-SE. The remaining 6 tRNA genes (tRNA-Val, tRNA-His, tRNA-Phe, tRNA-SerAGN, tRNA-Cys, and tRNA-Met) not identified in this manner, were assigned by comparison with the tRNA genes of A. armillatus (no. NC_005934.1). Two rRNA genes (rrnS and rrnL) separated by a tRNA-Val were identified in the mt genome, with the boundaries defined by contiguous genes (nad1 and tRNA-LeuUUR). The rrnS and rrnL were found to be 659 bp and 1,144 bp, respectively.
The major NCR of 2,769 bp found between tRNA-LeuUUR and tRNA-SerUCN, was smaller than that of A. armillatus (2,993 bp). This region could be divided into 3 parts, with the first being 837 bp in length and having an A+T content of 52.93%. The second region was the longest and contained 2 tandem repeats (1,496 bp in length) with 72.41% A+T nucleotides, while the third part was 62 bp in length and located before the tRNA-SerUCN gene.
Phylogenetic analysis based on 6 PCGs (atp6, cox1-3, and nad2), placed A. agkistrodontis and A. armillatus as a definitive cluster in the clade Pentastomida, grouped with Argulus americanus (Branchiura) in the clade Ichthyostraca [16]. The resulting phylogenetic tree also revealed that all the species in Arthropoda are distributed into 3 independent clades: Chelicerata; Mandibulata, and Pancrustacea (Fig. 2). This is consistent with the phylogenomic analysis of nuclear protein-coding sequences [17], in which this clade was named as Oligostraca to represent a subclass in the clade Maxillopoda (Branchiura, Pentastomida, and Mystacocarida) and clade Ostracoda. However, when we constructed a phylogenetic tree based on the analysis of 12 PCGs (except for atp8), the clade Ichthyostraca (containing Branchiura and Pentastomida) was located as a sister group with Chelicerata (Fig. 1S). We suspect this analysis is skewed by long-branch attraction bias caused by the noise introduced with the inclusion of these more rapidly evolving genes. For this reason, we suggest using the 6 most conserved PCGs (atp6, cox1-3, and nad2) be used to analyze the phylogenetic relationships among the Pentastomida within other species. While evidence from palaeontological [18–20], comparative spermatology [21–23], and molecular biomarkers [8,16] has been used to classify and group pentastomids, the actual evolutionary relationship between these species is, as yet, unresolved and remains controversial. Therefore, complete genome information from more pentastomid species is required to clarify this issue.
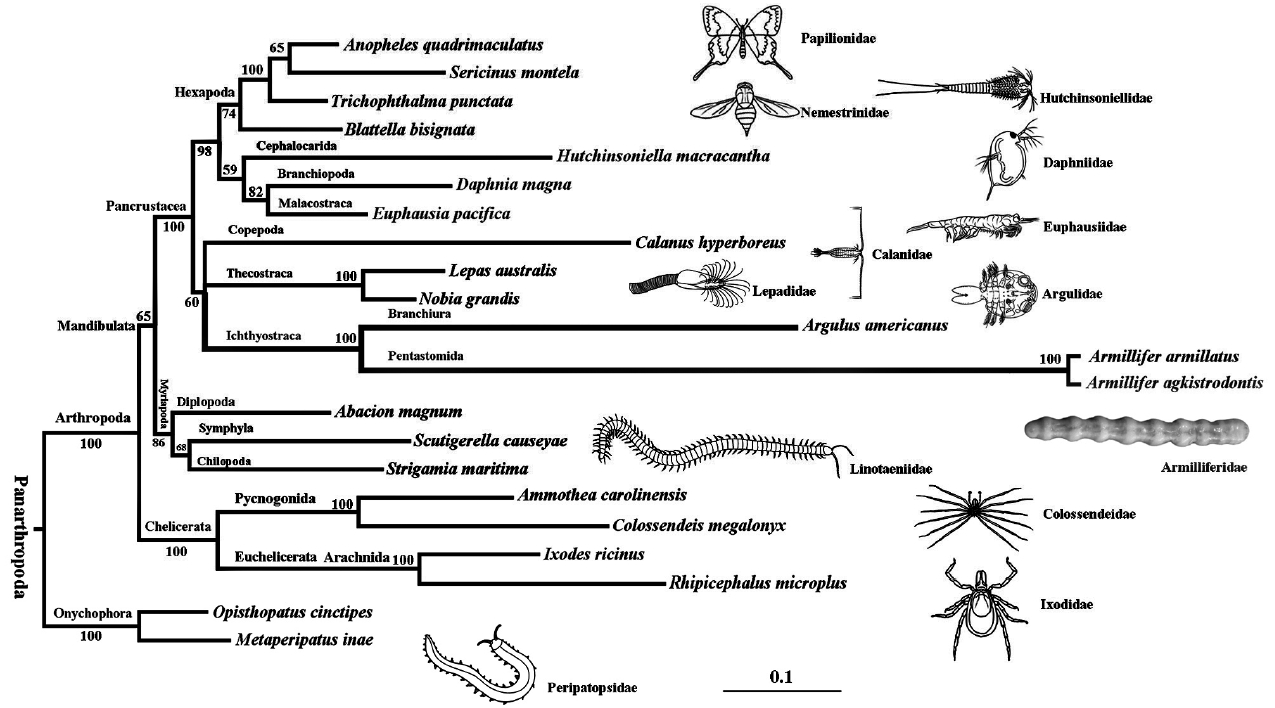
A phylogenetic tree of Armillifer agkistrodontis with other arthropods based on 6 conserved mitochondrial protein coding genes. Opisthopatus cinctipes and Metaperipatus inae (Onychophora) were used as outgroups.
In conclusion, this study presents the first complete mt genome sequence from A. agkistrodontis. Our phylogenetic analysis supports the previous observation that A. agkistrodontis has strong similarity to the Branchiura and should be grouped in the Crustacea. Finally, the data presented here have provided more molecular biomarkers for future studies of epidemiology, discrimination of cryptic species, and the analysis of genetic diversity, and systematic classification of pentastomids.
Supplementary materials
A phylogenetic tree of Armillifer agkistrodontis with other arthropods based on concatenated amino acid sequence data representing 12 mitochondrial protein coding genes. Opisthopatus cinctipes and Metaperipatus inae (Onychophora) were used as outgroups.

Organization of the mitochondrial genome of Armillifer agkistrodontis
ACKNOWLEDGMENT
This work was supported, in part, by the National Science and Technology Major Project (no. 2009ZX10004-302) to the corresponding author (Hu Wei).
Notes
CONFLICT OF INTEREST
There is no conflict of interest related to this work.