Genotypic Identification of Cystoisospora in Immunocompromised Patients Using Tm-Variation Analysis
Article information
Abstract
Cystoisospora is responsible for morbidity in immunocompromised patients. PCR is sensitive for diagnosing Cystoisospora; however, it needs reevaluation for differential molecular diagnosis of cystoisosporiasis. We aimed at evaluating melting curve analysis (MCA) after real-time PCR (qPCR) in diagnosis and genotyping of Cystoisospora as an alternative to conventional PCR. We included 293 diarrheic stool samples of patients attending the Department of Clinical Oncology and Nuclear Medicine of Cairo University Hospitals, Egypt. Samples were subjected to microscopy, nested PCR (nPCR), and qPCR targeting the internal transcribed spacer 2 region (ITS2) of the ribosomal RNA (r RNA) gene followed by melting temperatures (Tms) analysis and comparing the results to PCR-RFLP banding patterns. Using microscopy and ITS2-nPCR, 3.1% and 5.8% of cases were Cystoisospora positive, respectively, while 10.9% were positive using qPCR. Genotyping of Cystoisospora by qPCR-MCA revealed 2 genotypes. These genotypes matched with 2 distinct melting peaks with specified Tms at 85.8°C and 88.6°C, which indicated genetic variation among Cystoisospora isolates in Egypt. Genotype II proved to be more prevalent (65.6%). HIV-related Kaposi sarcoma and leukemic patients harbored both genotypes with a tendency to genotype II. Genotype I was more prevalent in lymphomas and mammary gland tumors while colorectal and hepatocellular tumors harbored genotype II suggesting that this genotype might be responsible for the development of cystoisosporiasis in immunocompromised patients. Direct reliable identification and differentiation of Cystoisospora species could be established using qPCR-Tms analysis which is useful for rapid detection and screening of Cystoisospora genotypes principally in high risk groups.
INTRODUCTION
Cystoisospora, formerly called Isospora, after taxonomic re-arrangements, were proved to infect human beings solely [1]. The display of cystoisosporiasis is inconsistent, depending on the parasite aggression as well as the host immune condition [2]. Diarrheal cystoisosporiasis may affect both immunocompetent and immunosuppressed individuals. However, it seems to occur more frequently in the latter. Thus, it was described as an opportunistic protozoan [3]. Despite that patients with cystoisosporiasis usually improve within a few days after commencement of treatments, relapses of diarrhea have been reported in a large number of cases with failing to spot Cystoisospora as the cause of recurrent diarrhea. This reinfection may be missed as oocysts have thin translucent walls and are usually scanty and intermittent in stool samples which in turn make microscopic examination results unremarkable [4].
Staining for detection of Cystoisospora, as auramine-O, reported 100% sensitivity and specificity compared to modified Zeihl-Neelsen stain, while iodine staining wet mount showed 54% sensitivity. However, repeated multiple testing using expertise microscopist is required [5]. As a result of the difficulty associated with identifying Cystoisospora in fecal samples, diseases allied with its genotypes are likely underdiagnosed, and their outburst reports may be underestimated. Subsequently, more sensitive tools for detection of Cystoisospora are inevitable especially for species identification. Cystoisospora-specific molecular analysis is accessible for much more sensitive screening [6].
Muller et al. [7] described nested PCR (nPCR) with southern blot hybridization for the detection of light C. belli infections. However, these are very lengthy and not competent for epidemiologic purposes. Moreover, PCR primers recognizing conserved DNA sequences of Cystoisospora were used in intestinal tissue biopsy and were confirmed at replicate tissue analysis [3]. Taniuchi et al. [8] eluded diagnosis using conventional methods by suggesting higher sensitivity of molecular approaches compared to microscopy. They reported that multiplex PCR detected C. belli in stools with 93% sensitivity compared to microscopy, and some of C. belli null samples tested positive by real-time PCR assay. Different molecular targets of protozoa could be used by real-time PCR assay for potent diagnosis in epidemiologic studies [9]. Real-time PCR with MCA of coccidian oocysts in different diagnostic samples could be sensitively used and it consistently discriminated different isolates [10]. Thus, herein we proposed a molecular diagnosis using qPCR-MCA for genotyping of cystoisosporiasis in immunocompromised patients with diarrhea to investigate this approach as a potential assay for genotyping Cystoisospora that may elude diagnosis using tedious conventional approaches.
MATERIALS AND METHODS
Patients
This study included a total of 293 diarrheic stool samples (198 males and 95 females). Patients’ ages ranged from 15 to 55 years with a mean age of 25.3 years; attending the Department of Clinical Oncology and Nuclear Medicine of Cairo University Kasr Al Ainy Hospitals from September 2015 to December 2016. Patients attended for diagnosis and treatment of various types of tumors, including HIV-related Kaposi’s sarcoma (3 patients), hematological malignancies, leukemias and lymphomas (134 patients), colorectal carcinomas (48 patients), hepatocellular tumors (76 patients), and mammary gland tumors (32 patients). All stool samples were examined in the research laboratory of Parasitology Department, Faculty of Medicine, Cairo University.
Direct microscopy of fecal smears
All samples were investigated by microscopic examinations of wet mounts. Stools were then concentrated by a formalinethyl acetate technique, stained using modified acid fast stain and microscopically examined using low-power (×10) and high-power (×40) objectives.
Specimens and DNA extraction
All stool samples were stored at -20°C for less than 6 months before DNA extraction. DNA was extracted using QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Potential inhibitors were removed by further purification with the QIA quick PCR purification kit (QIAGEN Inc.) according to the manufacturer’s instructions. Purified DNA was stored at −20°C until used.
nPCR of ITS2 rRNA gene
According to Taniuchi et al. [11] and Jongwutiwes et al. [2], a nPCR of the ITS2 rRNA gene included 2 consecutive PCR reactions. The first reaction employs an external pair of primer sets (Fermentas UAB, Vilnius, Lithuania) Iso-18SF0 (5′-TGGTTGATCCTGCCAGTA-3′) and Iso-28SR0, (5′-AAGGCTCAATCAAGAACCTCCG-3′) which amplify the DNA fragment spanning the SSU rRNA, ITS-1, 5.8S rRNA and ITS-2 regions of Cystoisospora. The second reaction contained 2 nested primers internal to the first primer pair and amplified a 404 bp fragment. These were Ib-213 F (5′-GGATATTCCCTGCAGCATGT-3′) and a reverse one: Ib-213R (5′-CGGGACACAACTCAACACTG-3′) (GenBank accession no. AF443614). Amplification was done using 12.5 μl PCR Master Mix (product no. B2281; Thermo Scientific, Runcorn, UK), 1 μl (200 nmol/L) of each forward and reverse primer, 2.5 μl of template DNA, 0.1 μl Taq polymerase (5 U/μl) (product no. EP3501, Thermo Scientific) and 8 μl of sterile distilled water to complete a total volume of 25 μl using thermal cycler (Biometra, Göttingen, Germany). The thermal cycling conditions were the same in both PCR reactions; denaturation at 94°C for 40 sec, annealing at 64°C for 40 sec, extension at 74°C for 5 min for 35 cycles of amplification. The amplified products were separated by electrophoresis on 2% agarose gel and visualized under a transilluminator after staining with ethidium bromide.
qPCR-MCA
It was carried according to Hove et al. [9] targeting ITS2 region of rRNA gene. The amplification was performed using the forward primer Ib-40F (5′-ATA TTC CCT GCA GCA TGT CTG TTT-3′) and reverse primer Ib-129R (5′-CCA CAC GCG TAT TCC AGA GA-3′), that amplified a 89 bp fragment of ITS2 region of rRNA gene. Double-labeled probe Ib-81Taq (FAM-5′-CAA GTT CTG CTC ACG CGC TTC TGG-3′-BHC1) (Invitrogen, Carlsbad, California, USA) were used. All qPCR assays were performed with the iCycler iQ real-time PCR detection system (Bio-Rad Laboratories, Hercules, California, USA). The final reaction mix contained 13 iQ SYBR Green Supermix (Invitrogen) 400 nM for R-Cystoisospora, F-Cystoisospora, and sterile dH2O adjusted to a final volume of 25 ml. Standard curves for all qPCR assays were prepared by 10-fold dilution of C. belli DNA. We used the standard curves to identify the reference and alternate samples. As the MCA method relies on saturating DNA dyes, the standard and variant samples produce single Tm peaks that diverge by 0.7–1.2°C [12]. This allowed us to identify genotypes by their single Tm peak and their association with the standard Tm curve with subsequent identification of genotypes by their shifted Tm curves compared to the standard curves. The PCR cycling conditions were 3 min at 95°C, followed by 40 cycles of denaturing at 95°C for 40 sec, annealing at 62°C for 40 sec, and extension at 72°C for 40 sec. After finishing of 40 cycles PCR amplification, the PCR products were melted by elevating the temperature from 40°C to 95°C at a rate of 1°C/min. Melting curves were generated by plotting for each sample the rate of change in fluorescence against the increase in temperature. The iCycler iQ software displayed the records collected during MCA as −dF/dT vs temperature. A sample was considered positive when the icycler determined a crossing point in the quantification analysis screen. Tms were detected by melting peaks of the curve.
Correlation of RFLP banding patterns to the corresponding Tms peaks
After the qPCR amplification of ITS2-rRNA gene and MCA, the PCR products belonging to the detected Tms were digested using RFLP with the enzyme AluI (Thermo Scientific, Waltham, Massachusetts, USA). The restriction digestion reaction consisted of a 10 Ul aliquot of PCR product, 1 U of AluI, 2 Ul of acetylated BSA (Thermo Scientific), and made up to a total volume of 30 Ul with nuclease-free water, then incubated in a 37°C water bath for 1 hr. Restriction fragments were separated on 2% agarose gels and visualized under UV illumination to observe different banding patterns and to compare these patterns with the corresponding Tms.
Specificity of qPCR assay
To determine the analytical specificity of the qPCR, DNAs were evaluated separately. We used in each run: (a) a distilled water sample as a negative control. (b) Cystoisospora control DNA extracted from an unpreserved human fecal sample in which the presence of Cystoisospora oocysts was confirmed by microscopy. (c) DNAs isolated from Schistosoma mansoni, Fasciola gigantica, Cyclospora cayetanensis, Entamoeba histolytica, Entamoeba dispar, Giardia lamblia, Cryptosporidium parvum, Enterocytozoon bieneusi, Encephalitozoon intestinalis. E. histolytica, E. dispar, and G. lamblia (n=10 for each).
Statistical analysis
Means (±SD) of the Tms in duplicate independent assays were calculated.
Ethical considerations
This study was conducted in compliance with the Helsinki Declaration. The research was approved by the Scientific Research Ethical Committee, Faculty of Medicine, Cairo University (archiving no. 15/2017). All procedures were explained to patients and a written or thumb printed consent was obtained.
RESULTS
Among the 293 diarrheic stool samples included in the present study, Cystoisospora isolates were detected in 9 cases (3.1%) and 17 cases (5.8%) by microscopy and nPCR, respectively. While by using qPCR, Cystoisospora was detected in 32 samples (10.9%) (Table 1). Using the ITS2-rRNA gene amplification by qPCR followed by MCA, we demonstrated variation in Tms between the standard curves and the tested samples. Two different Tms were found, including 85.8°C, 88.6°C. Each Tm corresponds to a specific distinct curve peak which in turn implies for a specific genotype, facilitating its identification (Fig. 1A–D). Consistent Tms were obtained for each genotype in duplicate independent assays with inter-assay variation less than 0.3°C (Table 2). As a result, 2 genotypes with 2 different peaks; genotype-I and genotype-II were found corresponding to 2 Tms; 85.8°C and 88.6°C, respectively. Among 32 individuals harboring Cystoisospora isolates, 21 were harboring genotype-II isolates (65.6%) while genotype-I was detected in 10/32 (31.3%), and there was 1 sample which harbored both genotypes. Genotype I was detected in 5 leukemic patients, 3 lymphoma patients, and 2 patients with mammary gland tumors. Genotype II was detected in 11 leukemic patients, 6 colorectal, 2 hepatocellular carcinoma (HCC), and 2 HIV-related Kaposi carcinoma. Both genotypes were detected in 1 HIV-related Kaposi carcinoma. By using restriction enzyme AluI for digestion of amplified qPCR products targeting the ITS2 gene, we found that DNA samples that melted at 85.8°C Tm resulted in DNA bands of 55 and 67 bp which corresponds to genotype I. Likewise, DNAs that melt at 88.6°C Tm resulted in different bands of 21 and 130 bp which correspond to genotype II (Fig. 2; Table 2). Diagnostic yields of qPCR-ITS2 showed the highest sensitivity of 100%, nPCR showed a sensitivity of 53.1%, while microscopy had the least sensitivity of 28.1%. All assays were 100% specific.

Diagnostic yield of microscopy, nPCR, and qPCR-ITS2 in the studied group
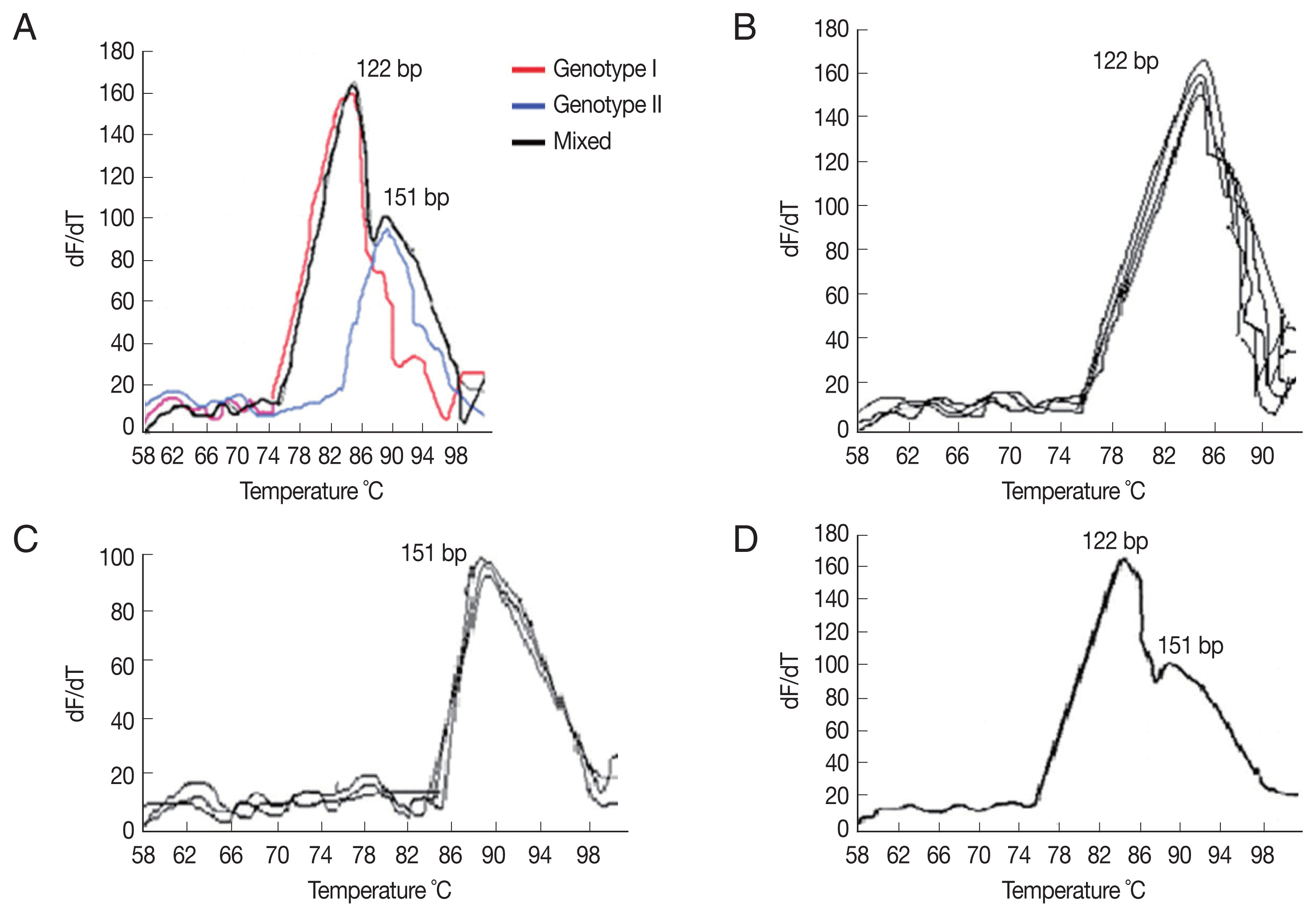
(A) Melting curve analysis of qPCR amplification products of Cystoisospora. The amplified DNA products represented as distinct melting peaks with specified Tms (88.6°C and 85.8°C). Each Tm corresponds to a specific genotype. (B) Melting curve of confirmed samples of Cystoisospora harboring genotype I showing distinct melting peaks at 85.8°C. (C) Melting curve of confirmed samples of Cystoisospora harboring genotype II showing distinct melting peaks at 88.6°C. (D) Melting curve of a confirmed sample of Cystoisospora harboring both genotypes II and I showing 2 distinct melting peaks at 88.6°C and 85.8°C, respectively.

Mean Tms of qPCR and RFLP product size of amplified Cystoisospora DNAs
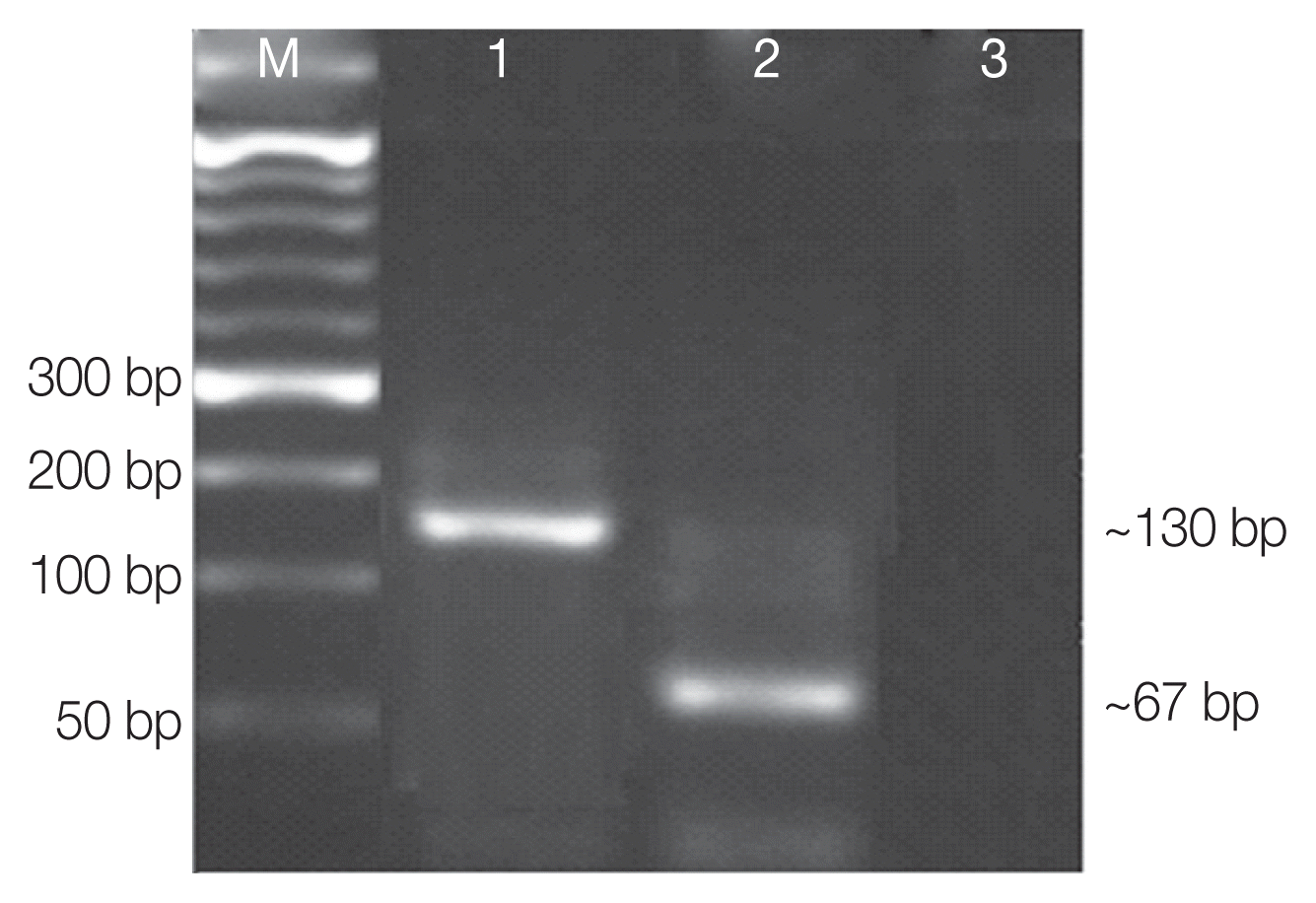
Agarose gel electrophoresis of RFLP profiles of ITS2 qPCR amplified Cystoisospora products using ALU I restriction enzyme. Lane M, 50 bp molecular weight marker; lane 1, confirmed sample (genotype II: 130 bp); lane 2, confirmed sample (genotype I: 67bp); lane 3, control.
DISCUSSION
Little is known about characterization of Cystoisospora genotypes from humans and animals [6]. In Egypt, cystoisosporiasis is diagnosed sporadically, with more prevalence among the immunocompromised patients, especially those with HIV and cancers. However, it may be under-valuated and most likely higher detection could be during routine investigations of target groups. In 2006, Isospora hominis have been documented in Egypt with a prevalence reaching 7.7% [13]. To address this concern, we employed the ITS2-rRNA gene amplification by q-PCR followed by MCA. During our search through literatures, few studies analyzed species/strain variation in Cystoisospora targeting the ITS2 rRNA gene [2,9,11].
ITS regions are non-coding areas among 28S, 5.8S, and 18S rRNA genes and are extremely preserved through diverse species. ITS2 sequence is considered a barcode as it is highly preserved, easy to be amplified, and allows high discrimination of closely related groups [14,15]. Consequently, the qPCR assay described in the current study allowed Cystoisospora diagnosis with improved sensitivity when compared to nPCR assay. Moreover, this qPCR has the prospective to reveal few oocysts shedding Cystoisospora patients as it depends on incorporating a specific intercalating dye; SYBR Green which is a double-stranded DNA binding dye that results in fluorescence emission curves when binding to the PCR product after each extension step. Thus, avoids electrophoresis of amplicons and allows quantitation through detection of the cycle (Ct) at which the amplification initiates [16]. Moreover, Ririe et al [17] reported the advantage of MCA over the conventional PCR that qPCR products can be differentiated during amplification by analysis of melting curves whose shape is a property of GC content, length and sequence. MCA allows differentiation of diverse genotypes relying on variations in their shapes and Tms shift as a result of variable bond strengths connecting base pairs [18]. Consequently, we interpret the results of the qPCR-MC depending on the Tms of the specific amplicons which were sufficiently different and constant to allow differentiation of the products. In our study MCA yielded 2 melting peaks (Tms), each corresponds to a specific genotype of Cystoisospora. Tms were consistent, with inter-assay variations less than 0.3 and were wide apart allowing easy discrimination between the different MCs. Genotype II belonging to 88.6°C Tm was more prevalent than the other genotype suggesting that this genotype is more responsible for cystoisosporiasis in Egypt. Notably, we found that qPCR-MCA achieved 100% sensitivity compared to microscopy (28.1%) and nPCR (53.1%). Both latter assays had 23 and 15 false negative cases, respectively, which were properly detected by qPCR. What is more, on comparing RFLP products to the corresponding Tms, we found 2 different banding patterns that match to the 2 variant Tms. Subsequently, these results confirm the genetic variation among Cystoisospora isolates, achieving analogous results to the qPCR-MCA and proving 100% sensitivity and specificity for qPCR-MCA.
In conclusion, our results highlighted Cystoisospora infection in Egypt with 2 distinct genotypes and specifically approaching immunocompromised patients. Moreover, ITS-2 rRNA-MCA offered rapid, sensitive, specific approach than nPCR as it overcomes post amplification manipulations, the need for restriction digestion study and further, qPCR offers easier interpretation of MCA results than visual assessment of stained gels. Accordingly, the potential of establishing qPCR-Tms analysis for rapid detection and screening of Cystoisospora genotypes in case of endemic population as routine screening of high risk groups should be considered.
Notes
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.