An Alternative Method for Extracting Plasmodium DNA from EDTA Whole Blood for Malaria Diagnosis
Article information
Abstract
Molecular techniques have been introduced for malaria diagnosis because they offer greater sensitivity and specificity than microscopic examinations. Therefore, DNA isolation methods have been developed for easy preparation and cost effectiveness. The present study described a simple protocol for Plasmodium DNA isolation from EDTA-whole blood. This study demonstrated that after heating infected blood samples with Tris–EDTA buffer and proteinase K solution, without isolation and purification steps, the supernatant can be used as a DNA template for amplification by PCR. The sensitivity of the extracted DNA of Plasmodium falciparum and Plasmodium vivax was separately analyzed by both PCR and semi-nested PCR (Sn-PCR). The results revealed that for PCR the limit of detection was 40 parasites/μl for P. falciparum and 35.2 parasites/μl for P. vivax, whereas for Sn-PCR the limit of detection was 1.6 parasites/μl for P. falciparum and 1.4 parasites/μl for P. vivax. This new method was then verified by DNA extraction of whole blood from 11 asymptomatic Myanmar migrant workers and analyzed by Sn-PCR. The results revealed that DNA can be extracted from all samples, and there were 2 positive samples for Plasmodium (P. falciparum and P. vivax). Therefore, the protocol can be an alternative method for DNA extraction in laboratories with limited resources and a lack of trained technicians for malaria diagnosis. In addition, this protocol can be applied for subclinical cases, and this will be helpful for epidemiology and control.
INTRODUCTION
Malaria remains a life-threatening disease. In 2015, the World Health Organization reported that the number of cases globally was estimated at 214 million, accounting for nearly 438,000 deaths [1]. For malaria diagnosis, various reports have shown that microscopy and RDTs have low sensitivity with the limit of detection as low as 100 parasites/μl [2–4]. Therefore, PCR-based methods, including semi-nested PCR (Sn-PCR), have been introduced to overcome these problems [5,6].
DNA extraction is an important step in molecular diagnostic approaches. Numerous protocols have been developed to isolate Plasmodium DNA from a variety of blood sources in order to obtain high-quality DNA suitable for downstream applications [7,8]. Commercial DNA extraction kits are commonly used to reduce PCR inhibition caused by hemoglobin in red blood cell [9,10]. However, these kits are not always available in low-resource settings. Another method for DNA extraction in routine use involves the application of phenol and chloroform [11]. However, the drawbacks of the phenol-chloroform-based method are that it is labor-intensive and time-consuming, with a high risk of contamination due to the large number of tube changing steps. In 1994, genomic DNA extraction by isopropanol fractionation with concentrated sodium iodide (NaI) and sodium dodecyl sulfate (SDS) was successfully introduced and performed in a single tube, which reduced the loss and fragmentation of DNA caused by pipetting of DNA during multiple tube changes [12]. The microwave irradiation based DNA extraction has been reported to be rapid, cost-effective and easy to perform with 10 μl of whole blood and the results showed the limit of detection of 5 parasites/μl using nested PCR technique [9]. The limitation of this method is the capacity of tubes to be used should not be less than 0.5 ml, as they can damage when irradiated and might be contaminated with biological materials [9], the lysate obtained from heating infected red blood cells with proteinase K and TE buffer was purified by isotachophoresis (ITP) prior to performing PCR and quantitative PCR. This method showed a limit of detection of 500 parasites/μl. Nevertheless, this technique is the high cost because of using the specialized device, microfluidic, for DNA purification.
Therefore, the aim of this study was to demonstrate a simple protocol using TE buffer and proteinase K to extract Plasmodium DNA from EDTA-whole blood for subsequent downstream applications directly, without prior purification methods. The sensitivity of the established method was determined by serial dilution with normal blood and analysis using PCR and Sn-PCR. In the present study, this new protocol was verified by DNA extraction from whole blood of asymptomatic migrant workers who attended the screening program in order to work in Chiang Mai Province. In addition, the steps for extraction of the new protocol were compared with a reference method, commercial DNA extraction kit, and a previously published DNA extraction protocol [12] by focusing on cost-effectiveness and labor-intensiveness.
MATERIALS AND METHODS
Whole blood samples were obtained from patients at a hospital in Chiang Mai province, Thailand. EDTA-treated samples were examined for P. vivax and P. falciparum by microscopy; parasitemia levels in the blood samples were 0.38% and 0.01%, respectively. Normal blood taken from healthy persons with no history of malaria infection was serially diluted with malaria-infected blood. To verify the new method, whole blood samples obtained from 11 asymptomatic Myanmar migrant workers who attended screening program in order to work in ChaingMai Province. After routine diagnosis, the samples were transported to the laboratory in a cooler and then stored at −20°C until used. The study protocol was approved by the Khon Kaen University Ethics Committee for Human Research (HE591044). Informed consent was obtained from the study subjects using a standard approved procedure.
P. falciparum and P. vivax DNA was extracted from EDTA-treated whole blood samples using 3 methods: a commercial DNA extraction kit (QIAamp DNA Blood Mini Kit, Qiagen, Hilden, Germany); a previously published DNA extraction method [12] and the new protocol (TE buffer and proteinase K-based extraction). The protocols for DNA extraction by the 3 methods are given in Table 1. For Polymerase chain reaction, each gene of P. falciparum and P. vivax DNA was performed in separate PCR reactions using specific primers designed from 18S rRNA, as previously described [13]. A forward universal primer (UF) was designed from a genus-specific region, and reverse primers (FR for P. falciparum and VR for P. vivax) were designed from a species-specific region of P. falciparum and P. vivax. The oligonucleotide primers were as follows: UF, 5′-TCAGCTTTTGATGTTAGGGTATT-3′; FR, 5′--GCATCAAAGATACAAATATAAGC-3′; VR, 5′-TAAACTCCGAAGAGAAAATTCT-3′. The PCR tube contained a total volume of 20 μl, consisting of 10 mM KCl buffer, 5 mM Tris–HCl, 2.5 mM MgCl2, 200 mM dNTP, 0.5 μM of each primer, and 0.1 units of RBC Taq DNA polymerase (RBC Bioscience, New Taipei City, Taiwan). Reactions were performed in a GeneAmp PCR System 9700 thermal cycler (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The cycling conditions for P. falciparum and P. vivax consisted of an initial denaturation phase at 94°C for 2 min, 40 repetitions at 94°C for 30 sec, annealing of each primer pair at 47°C for 1 min, and extension at 72°C for 1 min, followed by a final extension for 10 min at 72°C. The PCR products were visualized by electrophoresis on 1.5% agarose gel stained with ethidium bromide.

DNA extraction methods from EDTA whole blood
To determine the limitations of DNA extracted by the new method, 2 malaria blood samples with known parasitemia, 0.38% for P. vivax and 0.01% for P. falciparum, were serially diluted with uninfected blood. The DNA extracted from each dilution was used as a template for conventional PCR and Sn-PCR. For conventional PCR, the conditions for PCR amplification were as described above (UF-VR for P. vivax identification, UF-FR for P. falciparum identification). The specific primers for Sn-PCR used in this study were designed from a small subunit of the Plasmodium ribosomal gene (ssrDNA), as previously described [14]. The primers are shown in Table 2. Sn-PCR amplification of each gene was performed separately, as follows. Nest 1: a total of 20 μl of mixture contained 10 mM KCl buffer, 5 mM Tris–HCl, 2.5 mM MgCl2, 200 mM dNTP, 0.5 μM of each primer, 0.1 units of RBC Taq DNA polymerase and 2 μl of DNA template. Nest 2: a total of 20 μl of mixture contained 10 mM KCl buffer, 5 mM Tris–HCl, 2.5 mM MgCl2, 200 mM dNTP, 0.5 μM of each primer, 0.1 units of RBC Taq DNA polymerase and 2 μl of 1:10 diluted DNA from the first reaction. All reactions were performed in a GeneAmp PCR System 9700 thermal cycler. The amplicon was visualized by 1.5% agarose gel electrophoresis and stained with ethidium bromide. To verify the new method, the EDTA-whole blood of 11 asymptomatic Myanma migrant workers who attended the screening program at ChaingMai hospital were used to extract DNA by using the new protocol and analyzed by Sn-PCR, using specific primers as shown in Table 2. The first reaction, which included primers UNR, PLF and HUF, used a total of 20 μl of a mixture consisting of 10 mM KCl buffer, 5 mM Tris–HCl, 2.5 mM MgCl2, 200 mM dNTP, 0.1 units of RBC Taq DNA polymerase, and 2 μl of DNA template. The amounts of the 3 primers used in this reaction were 0.5 μM for UNR, 0.5 μM for PLF, and 0.05 μM for HUF. The second reaction, which included PLF, FAR and VIR, used a total of 20 μl of a mixture consisting of 10 mM KCl buffer, 5 mM Tris–HCl, 2.5 mM MgCl2, 200 mM dNTP, 0.5 μM of each primer, 0.1 units of RBC Taq DNA polymerase, and 2 μl of 1:10 diluted DNA from the first reaction. All reactions were performed in a GeneAmp PCR System 9700 thermal cycler. The amplicon was visualized by 1.5% agarose gel electrophoresis and stained with ethidium bromide. The estimated cost for each of the 3 methods used to extract DNA from the samples was calculated based on the purchase price of the reagents and kits that were used in US dollars (USD).

List of primers for semi-nested PCR (Sn-PCR) used in this study
RESULTS
P. falciparum and P. vivax DNA extracted from EDTA whole blood using the new method (TE buffer and proteinase K-based extraction) was successfully amplified by PCR as shown in Fig. 1. The product sizes of the target genes of P. falciparum was 1,451 bp and P. vivax was 833 bp. PCR products were not obtained from the negative control lacking Plasmodium species. This negative result indicates that the reagents had not been contaminated while setting up the process. The sensitivity of PCR and Sn-PCR assays after TE buffer and proteinase K-based extraction was determined based on serial dilution of EDTA-treated whole blood with normal blood. P. falciparum was diluted from 1,000 to 0.06 parasites/μl, and P. vivax was diluted from 4,400 to 0.06 parasites/μl. Standard PCR was then performed using specific primers, as previously described [13]. The results revealed that the limit of detection was 40 parasites/μl for P. falciparum and 35.2 parasites/μl for P. vivax (Figs. 2, 3). For Sn-PCR assay, P. falciparum was diluted from 5,000 to 0.002 parasites/μl, and P. vivax was diluted from 22,000 to 0.01 parasites/μl. The results revealed that the limit of detection was 1.6 parasites/μl for P. falciparum and 1.4 parasites/μl for P. vivax (Figs. 4, 5).

Amplified P. falciparum and P. vivax gene products after PCR. (A) PCR product of P. falciparum. (B) PCR product of P. vivax. Lane M, DNA ladder; lane C, negative control; lane 1, PCR product of DNA extracted using a NucleoSpin® Blood; lane 2, PCR product of DNA extracted by a published DNA extraction method; lane 3, PCR product of DNA extracted by the modified method (TE buffer and proteinase K-based extraction). The arrow indicates the expected size of amplicons.

Limit of detection of P. falciparum using TE–proteinase K-based method and standard PCR. The numbers above each lane indicate the number of parasites present per μl of blood. Infected whole blood was serially diluted with normal whole blood. The initial parasitemia of P. falciparum was 0.01%. Lane M is a DNA size marker.

Limit of detection of P. vivax using TE–proteinase K-based method and standard PCR. The numbers above each lane indicate the number of parasites present per μl of blood. Infected whole blood was serially diluted with normal whole blood. The initial parasitemia of P. vivax was 0.38%. Lane M is a DNA size marker.

Limit of detection of P. falciparum using TE–proteinase K-based method and semi-nested PCR. The numbers above each lane indicate the number of parasites present per μl of blood. Infected whole blood was serially diluted with normal whole blood. The initial parasitemia of P. falciparum was 0.01%. Lane M is a DNA size marker. (A) The first PCR reaction showed a limit of detection of 40 parasites/μl. (B) The second PCR reaction showed a limit of detection of 1.6 parasites/μl.

Limit of detection of P. vivax using TE–proteinase K-based method and semi-nested PCR. The numbers above each lane indicate the number of parasites present per μl of blood. Infected whole blood was serially diluted with normal whole blood. The initial parasitemia of P. vivax was 0.38%. Lane M is a DNA size marker. (A) The first PCR reaction showed a limit of detection of 35.2 parasites/μl. (B) The second PCR reaction showed a limit of detection of 1.4 parasites/μl.
The new protocol showed successful DNA extraction from EDTA whole blood of 11 asymtomatic Myanmar migrant workers and then analyzed by Sn-PCR as shown in Fig. 6. The product size of the human gene from the first reaction amplified from UNR and HUF primers was 231 bp (Fig. 6A). Thereafter, the amplicon from the first reaction was diluted 1:10 and used as template for the second reaction. The results of the second reaction which using specific primers for P. falciparum and for P. viax showed that there were 2 positive samples for P. falciparum at the target size of 395 bp (Fig. 6B, lane 7) and 499 bp for P. vivax (Fig. 6B, lane 2). Cost per sample was estimated based on the price of reagents or kits used in each of the 3 methods. For the commercial kit, the costs of the previously published DNA extraction method and the method established here were approximately 2.86 US$, 1.86 US$ and 0.15 US$, respectively. For time consuming, the commercial kit took around 4 hr and the published DNA extraction method took time around 2 hr and 30 min, while the new method took time to perform around 1 hr and 30 min. The new method here was, thus, more cost-effective and less time consuming than the 2 with which it was compared.
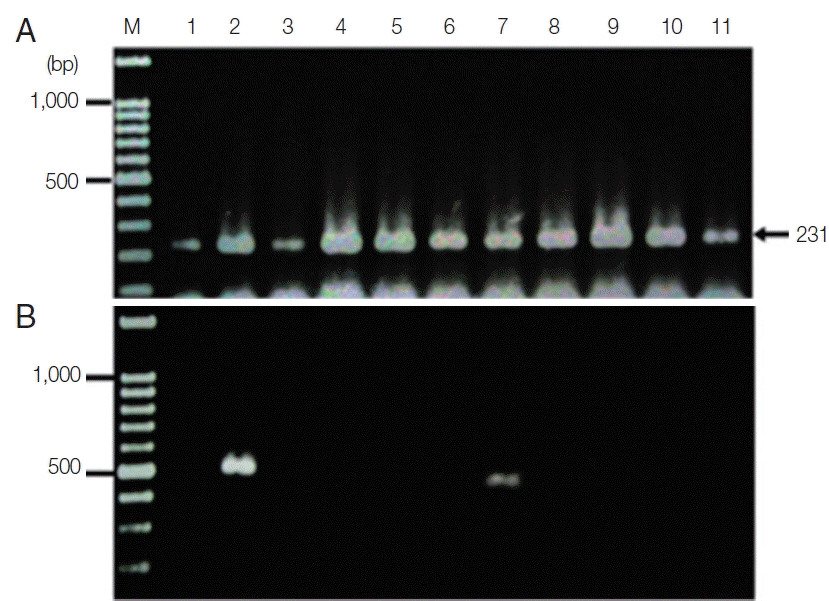
Agarose gel electrophoresis of the amplification products of the DNA template extracted using the TE–proteinase K-based method and semi-nested PCR. Lane M is a DNA size marker. Lanes 1–11 were amplicons from 11 asymptomatic migrant workers. (A) The first PCR reaction showed a positive DNA band amplified by UNR-HUF primers with the size of products at 231 bp. (B) The second PCR reaction showed P. vivax detection at lane 2 with a product size of 499 bp and P. falciparum at lane 7 with the target gene at 395 bp.
DISCUSSION
Over the years, multiple protocols have been developed and applied for extraction of Plasmodium DNA for various purposes, such as diagnostic [15,16], therapeutic uses [17], and epidemiology and control [18]. The aim of this study was to establish a simple protocol for DNA extraction from EDTA whole blood for malaria diagnosis. The protocol consisted of hemolysis by sterile distilled water and partitioning of the DNA from other cell components by incubating at 56°C after adding TE buffer and proteinase K solution. Following enzyme inactivation and centrifugation, the supernatant was directly used for malaria detection by molecular methods, and no chemicals were required for isolation or purification. This study demonstrated that Plasmodium DNA was successfully extracted using this protocol and could be subjected to both standard PCR and Sn-PCR amplification. The sensitivity level of DNA extracted by TE buffer and proteinase K-based method was determined from serial dilutions of blood samples. The results of standard PCR showed a limit of detection of 40 parasites/μl for P. falciparum and 35 parasites/μl for P. vivax. Sn-PCR technique had greater sensitivity, with a limit of detection of 1.6 parasites/μl for P. falciparum and 1.4 parasites/μl for P. vivax. Thus, the new protocol in this study could be used to extract Plasmodium DNA and apply diagnostic measures to overcome the deficiencies of identification by microscopic examination by using standard PCR or Sn-PCR. Moreover, this protocol was also verified by extraction DNA from EDTA whole blood samples of asymptomatic Myanmar migrant workers who attend the screening program in order to work in Chiang Mai Province and the results demonstrated that this protocol can be used to detect low parasite levels by Sn-PCR and there has been reported that this molecular technique can be applied successfully in cases of asymptomatic infection [14,19]. This will be helpful for epidemiology and control, since asymptomatic persons play an important role in the transmission of malaria,
Techniques that have been employed for malarial DNA extraction from whole blood include rapid boiling, Chelex-100 chelating resin, microwave irradiation, isotachophoresis, and boil and spin method [20]. This is the first study to only use TE buffer and proteinase K for Plasmodium DNA extraction from EDTA whole blood. The steps for DNA extraction in this protocol were compared with a commercial DNA extraction kit and a previously published DNA extraction method [12], as shown in Table 1. In addition, these 2 features are clear advantages of this protocol, which is easy to perform, cost-effective, and less labor-intensive than alternative methods. However, the DNA quality may deteriorate during long-term storage because the sample is relatively unpurified. The effective implementation plans for malaria control are mass testing and prompt treatment. Therefore, governments of the countries should provide funding for malaria control by using effective interventions. As mentioned above, microscopic examination is low sensitivity and need experienced technician to read blood film, molecular techniques have been used to address these problems. The new protocol described offers a simple and cost effective for DNA extraction that can be used for mass testing in malaria control.
The new protocol in this study offers simple, low cost and not labor-intensive. It can be an alternative method for DNA extraction from EDTA whole blood in a clinical laboratory of a hospital with limited resources and lacking an expert microscopist. In addition, this protocol can be applied for detection of low parasitemia or subclinical individauls by using Sn-PCR. This will be helpful for active malaria surveillance.
ACKNOWLEDGMENTS
This study was supported in part by grants from the Invitation Research Fund, Faculty of Medicine, Khon Kaen University (no. IN59208), and the Commission on Higher Education, Ministry of Education, Thailand.
Notes
The authors declare that there is no conflict of interest.