INTRODUCTION
Angiostrongyliasis is caused by the rat lung worm, Angiostrongylus cantonensis, a kind of nematode that was first identified and reported by Chen at 1935 in wild rats in the vicinity of Canton in China [1]. Humans accidentally acquire infection with this nematode either by eating poorly cooked intermediate hosts (freshwater snails and slug) or carrier hosts (land crabs and freshwater shrimps) or by consuming the contaminated vegetables containing third-stage larvae [2–7]. The definitive hosts of A. cantonensis are divided into permissive (rat) and nonpermissive hosts (mice, guinea pigs, rabbits, rhesus monkeys, and humans) based on whether the worms invade the lung and eventually attain sexual maturity. In the permissive host, the worms can migrate to lungs without causing severe eosinophilic meningitis. But in nonpermissive hosts, larvae generally fail to migrate to the lungs and terminate their development at a young adult worm stage dwelling in CNS until death, which can cause meningitis or meningoencephalitis characterized by obviously increasing eosinophil infiltrations [8,9].
Clinical exploration revealed that A. cantonensis invading human CNS caused eosinophilic meningitis and meningoencephalitis, and resulted in a serial of neurological symptoms, including persistent headache, paresthesia or hyperesthesia, nuchal rigidity, seizure, cognitive dysfunction, ataxic gait, and even unconsciousness [10–18]. These neurological symptoms were also identified in animals with A. cantonensis infection, which obviously presented lessening of the spontaneous activity and dysfunction for symmetry, balance and coordination of movements in infected rodents [19–21]. Further comparison of the pathological injuries indicated that eosinophilic response and tissue damage were more serious in nonpermissive hosts than in permissive hosts [22–27]. However, the potential molecular mechanism of the behavioral dysfunctions and neuronal injuries with A. cantonensis infection are still incompletely known.
To primary exploration of the molecular mechanismof pathological basis of the behavioral dysfunctions, the behavioral test, classical histological and immunohistochemical techniques, and transcriptomic analysis were conducted. Moreover, we mainly focused on the time points at 2, 3 and week 4 PI because the larvae are first detected in the cranial cavity of rodents at week 2 PI and their number peaked at week 3 PI. Thus, this study will provide the evidences for further research on pathological mechanism and the potential direction for clinical therapy.
MATERIALS AND METHODS
Experimental animals
Healthy female Sprague-Dawley (SD) rats and BALB/c mice aged between 6 and 8 weeks were supplied from the Center of Animal Experiments of Sun Yat-sen University, Guangzhou, P. R. China (SYSU, ZSSOM; permit no: 2016-122). All animals were housed in a specific pathogen-free environment, and the procedures done were strictly followed the Guide for the Care and Use of Experimental Animals of the National Institutes of Health, China. Both mice and rats were randomly divided into 4 groups (10 animals per group) as the control, week 2 PI, week 3 PI, and week 4 PI, and for each rat, 100 of the third-stage larvae (L3) of A. cantonensis were used to infect mice by the gavage method and 20 for each mouse according to our previous study [27].
Worm preparation
The L3 larvae of A. cantonensis were obtained from Biomphalaria glabrata, the intermediate host maintained in our laboratory for the life cycle of A. cantonensis. L3 larvae of A. cantonensis were harvested using the method described by Chen and Lai [28] and Lan and Lai [29]. Briefly, B. glabrata were homogenized and digested in a pepsin-HCl solution (pH 2.0, 500 IU pepsin/g tissue) at 37°C for 2 hr. The L3 larvae in the sediment were identified and collected.
Behavioral testing of balance beam
The balance beam task followed the modified version of Shear et al. [30] and Chan et al. [31]. Experimental rodents were trained to travel from the end of a narrow-suspended beam into a dark goal box. The number of foot slips and the time of completing trial were recorded by 2 observers. A foot slip was defined as a paw descending below the surface of the beam. The record of foot slips and time was started when the front paws crossed the starting position and ended once the nose of the experimental rodents entered the dark goal box. Animals were tested 3 times daily, for 3 consecutive days at week 2, 3, and 4 PI. If the animal fell from the beam, or took more than 3 min, the task was recorded as incompletion.
Behavioral testing of Morris water maze task
In Morris water maze task (MWMT), following the protocol described previously [34,35], the experimental rodents were released (not dropped) into the water in 4 random starting positions (N, S, E, and W) with the faces to the tank wall. Trial was started at the moment that the rodents were released and stopped when they reached the platform hidden beneath the surface of the water. Each trial limited 2 min for rats and 1 min for mice. The experimental rodents were guided to the platform when they could not find it within time limit and were allowed to stay on it for 30 sec before the next swim. All swimming tracks were recorded by computer tracking program (Ethovision Software, Noldus, Wageningen, The Netherlands).
Given 4 trails daily, rodents were trained for 3 consecutive days at week 2, 3, and 4 PI and 1 probe trial (2-min for rats and 1-min for mice) were given after 24 hr following the last acquisition trail. Latencies (spatial learning) and percentage of time stay in the target quadrant compared with the other quadrants (probe trial) were measured.
Transcriptomic data analysis
In our previous study, RNA libraries were constructed from mouse brain tissues collected on day 2 PI and week 1, 2, and 3 PI, after total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, California, USA). Similarly, the samples from the control mice were collected on the 21st day PI, and the samples were frozen in liquid nitrogen. The overall flow of RNA library construction and deep sequencing had been shown in our previous study [36].
The raw data was processed with a bioinformatics’ pipeline of filter low quality tags, trim adaptor, clean reads mapping to the Mus musculus genome using TopHat, Refseq genes expression analysis using cufflinks, annotate and classify RNAs into different categories. Expression patterns of apoptotic genes in the apoptotic pathways were analyzed in mice brain with A. cantonensis infection.
Histochemistry examination of neuronal injury
Animals were anesthetized with chloral hydrate (350 mg/kg) and perfused transcardially with 300 ml of 0.9% sodium chloride and then 400 ml of 4% paraformaldehyde (in 0.1 M phosphate buffer PB, pH 7.4). The brains of the control and week 2, 3, and 4 PI animals were collected, used for pathological examinations, postfixed in 4% paraformaldehyde (in 0.1 MPB, pH 7.4) overnight, successively immersed in 10%, 20%, 30% sucrose solution with PB until the tissue sank, and then sliced in 30 μm thick coronal sections on a semiconductor freezing microtome (Leica, Bensheim, Germany). The sections were stained with hematoxylin and eosin (HE) according to the conventional staining methods [37].
Immunohistochemistry examination of neuronal injury
Brain sections were pretreated with 0.3% H2O2 in 0.01M PBS at 37°C for 30 min. To process for conventional immunohistochemical staining, series of sections were incubated for 48 hr at 4°C with primary antibody: neuronal nuclei (a neuronal marker, mouse anti-NeuN 1:500 Chemicon, Temecula, California, USA), glial fibrillary acidic protein (an astrocyte marker, mouse anti-GFAP 1:500 Chemicon), ionized calcium binding adapter molecule 1 (a microglia and macrophage maker, rabbit anti-Iba1 1:500) and caspase-3 (a marker of apoptotic signal, rabbit anti-caspase-3 1:250), separately. After rinsing 3 times with PBS, sections were incubated with secondary antibody: anti-mouse IgG or anti-rabbit IgG (1:100, Sigma, St. Louis, Missouri) for 3 hr, respectively, and then with the corresponding mouse or rabbit PAP complex (1:200, Sigma) at room temperature for 2 hr. The DAB-peroxidase reaction (0.05% in 0.01 M PBS, pH 7.4, Sigma) was performed for 2–8 min and mounted onto gelatin-coated slides, dried, dehydrated, cleared with xylene, and covered with neutral balsam.
For immunofluorescence staining, the brain sections were incubated with rabbit anti-ECP (eosinophil cationic protein, a basic protein located in the eosinophil primary matrix, 1: 200, Invitrogen), mouse anti-NeuN, and rabbit anti-Caspase-3, respectively. Each sections were washed 3 times for 5 min in PBS and followed by 2 hr incubation with the secondary antibody: Alexa 594-conjugated goat anti-rabbit IgG (1: 300, Invitrogen) or Alexa 594-conjugated goat anti-mouse IgG (1:300, Invitrogen) at room temperature. After washing with PBS, the sections were mounted on gelatin-coated slides, cover slipped with antifade medium (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA) and observed under the fluorescence microscope (Carl Zeiss, Jena, Germany).
Western blotting of caspase-3 expression
Cold PBS-washed different brain tissues were collected and then blended with Bullet Blender (Next Advance, Inc. New York, USA) lysing in precooled cell lysis buffer. The mixtures were kept cool on ice for 30 min. The supernatants were recollected, after centrifuged at 12,000 rpm for 5 min at 4°C, for total protein content assay. Quantitative 30 μg of protein were inactivated and then resolved by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes, which were then blocked in 5% skim milk for other 2 hr followed by blocking with the rabbit anti-caspase-3 antibody or rabbit anti-cleaved-caspase-3 antibody (1: 200,) at 4°C overnight. Thereafter, the membranes were washed 3 times with PBST and incubated with HRP-conjugated goat anti-rabbit IgG secondary antibody (1:500) at room temperature for 90 min. Washed 3 times with PBST, the membranes were followed by detection with chemiluminescence and quantified using ChemiDoc Touch System (BioRad, Hercules, California, USA). The immunoreactive bands were normalized with respect to β-actin.
RESULTS
Worsened motor dysfunction of mice induced by A. cantonensis infection
Comparison of larval and neuronal injuries in both rodents showed that the percentage of larvae in the cerebrum was significantly higher in rats than in mice. In contrast, the percentage of larvae in the brainstem was significantly higher in mice than in rats. The percentage of larval distribution in the cerebellum was significantly higher in mice than in rats at week 2 and 3 PI. Besides, the recovery rate at week 3 PI was apparently higher compared to other infection stages (Supplementary Table 1).
Behavioral changes of rats and mice with A. cantonensis infection were detected to evaluate the neuronal injury. The results of balance-beam task showed that movement and motor coordination functions were damaged in both A. cantonensis-infected rats and mice. The infected rats and mice needed longer time to complete the task than the control after week 2 and 3 PI, respectively (Fig. 1A; Supplementary Table 2). Similarly, the times of foot slip were significantly increased in the infected rats and mice after week 2 and 3 PI, respectively (Fig. 1B; Supplementary Table 3). The results of grip strength task showed no significant difference in the rats and mice at the week 1 PI, but the hang time slightly decreased in rats after week 3 PI, and dramatically decreased in mice after week 2 PI (Fig. 1C; Supplementary Table 4). Thus, mice were earlier than rats to reveal the behavioral dysfunction; moreover, mice had the higher intensity of behavioral dysfunction than in rats.
A. cantonensis infection caused cognitive impairments in mice
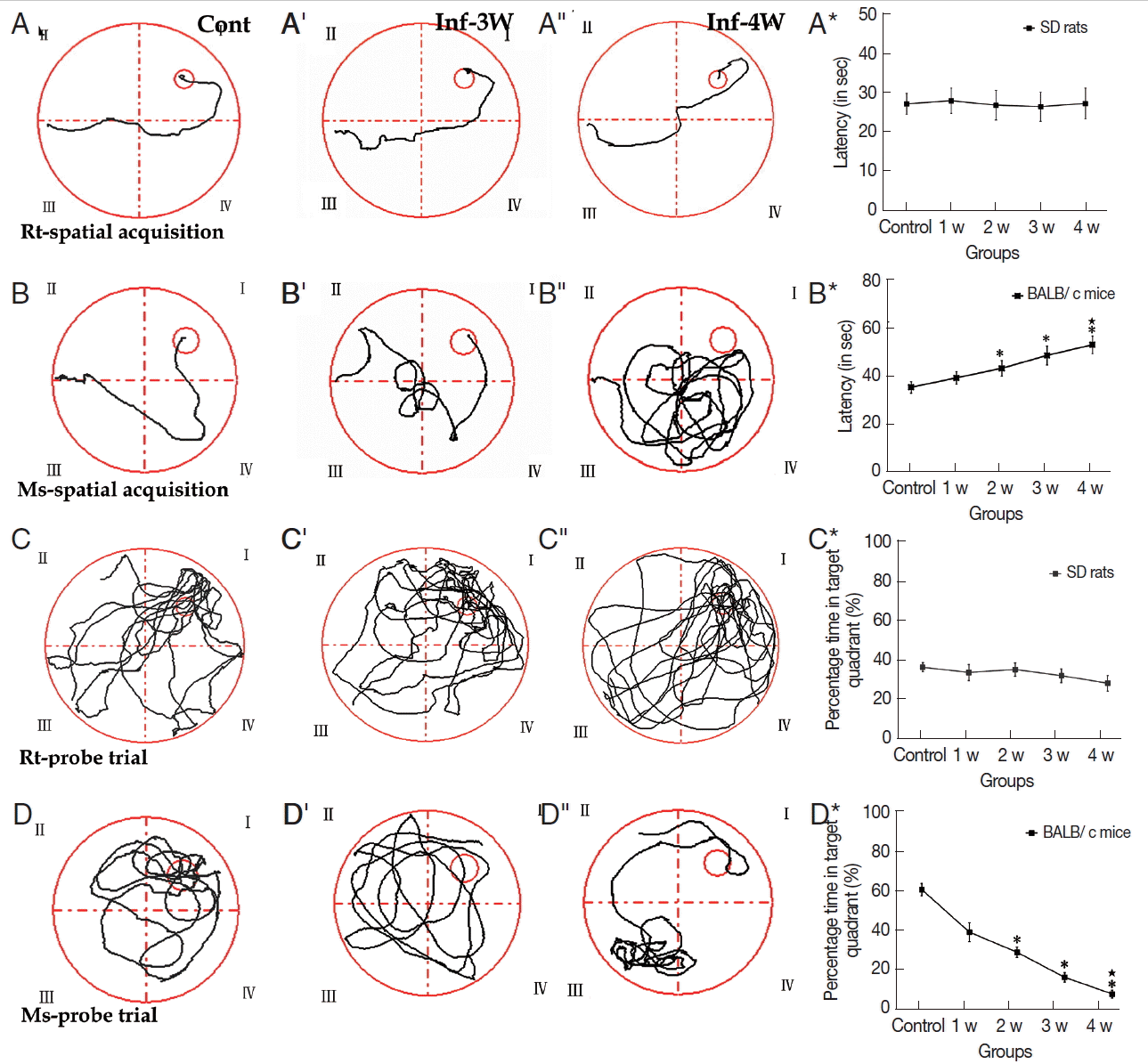
Assessments of cognitive impairments are the typical method to evaluate the neuronal injury of rodents with A. cantonensis infection. MWMT experimental data showed that no significant difference was observed in both tests of spatial learning and memory ability in rats (Fig. 2A-A*; Supplementary Table 5). In the spatial learning task, however, cognitive impairments became more serious compared to the control in the infectious course, and the infected mice needed more time to locate the hidden platform than the controls after week 2 PI (Fig. 2B-B*; Supplementary Table 5). In the percentage of time spent in the target quadrant of probe trail task, no significant change was observed in rats (Fig. 2C-C*; Supplementary Table 6), but the infected mice had lower levels compared with the control, and the lowest level appeared at week 4 PI (Fig. 2D-D*; Supplementary Table 6). Thus, cognitive impairments in mice, including sense, motor, learning, and memory, was more serious than in rats with A. cantonensis infection. The behavioral dysfunctions are consistent to the pathological injuries of the specific functional regions of the brain, including the cortex (sense and motor) and hippocampus (learning and memory). Therefore, our further experiments focused on these 2 regions in both rodents with A. cantonensis infection.
Difference in injuries between cerebral cortex and hippocampus in A. cantonensis infected rats and mice
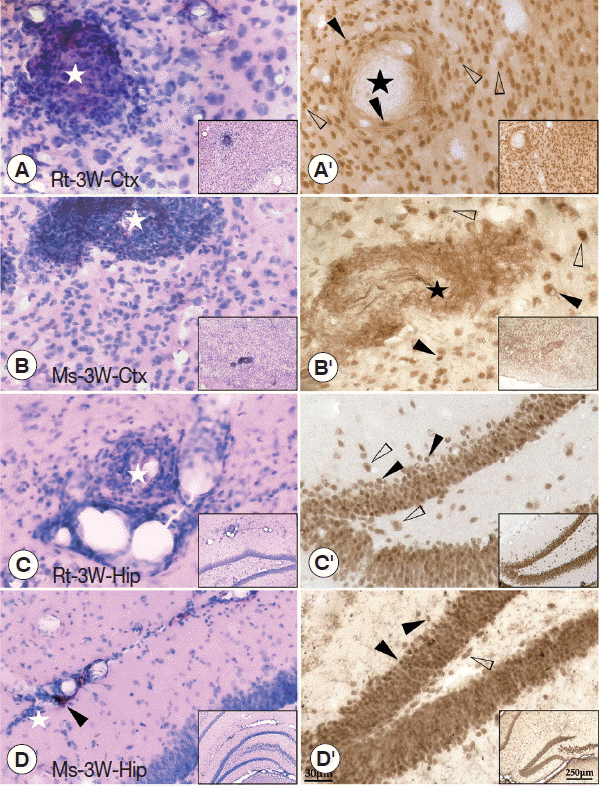
The pathological characters in the brain tissue of the cerebral cortex (sense and motor) and hippocampus (learning and memory) were carefully investigated at week 3 PI because serious behavioral dysfunctions could be found in rats at week 3 PI. The results showed that dilated vessels and perivascular cuffing of inflammatory cells were mainly observed in the cerebral cortex and hippocampus of mice and rats. Thus, pathological injuries in both regions were consistent to the behavioral dysfunctions in both rodents with A. cantonensis infection. In detail, plaque-like lesions were clearly observed near the dilated vessels in the cerebral cortex and hippocampus of both rodents with A. cantonensis infection (Fig. 3A–D). Immunohistochemical staining of NeuN showed that plaque-like lesions in the cerebral cortex were intensive with loss of neurons in the core (black star) (Fig. 3A′, 3B′). However, in the hippocampus, the blebbing in neurons, nucleus condensing (pyknosis), and cell shrinkage (loss of endochylema) (black arrowheads) could be found along with the hippocampal dentate gyrus and CA1 area (Fig. 3C′, 3D′), suggesting that the neuronal injuries accompanied with cell apoptosis in the hippocampus. Thus, the molecular mechanism of pathological damages between cerebral cortex and hippocampus might be different.
Extrinsic apoptosis pathway activated in the brain of A. cantonensis-infected mice
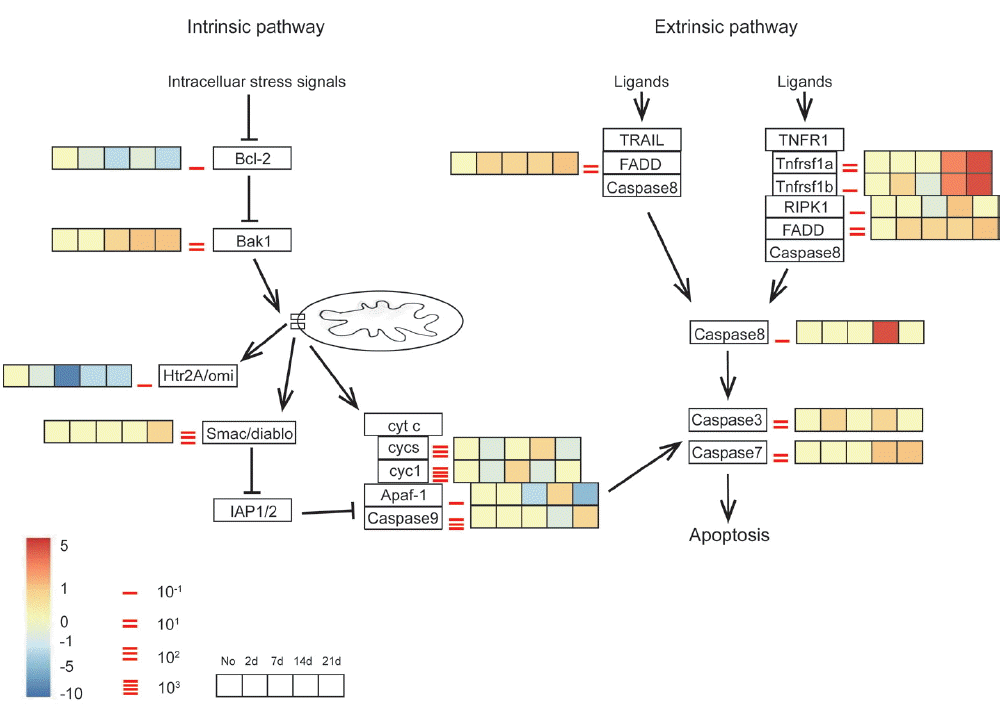
We further investigated expression patterns of the genes in the apoptosis pathways to explore the mechanisms of serious neuronal injuries in mice with A. cantonensis infection. In mammals, neuronal apoptosis is one of the typical reasons of pathogenesis of memory loss and cognitive impairment. Neuronal apoptosis can be initiated by 2 dominant pathways: (i) intrinsic pathway, which is triggered by cellular stress and the subsequent Bcl-2-stimulated the release of mitochondrial cytochrome c that is related to caspase-9 and Apaf-1 activation; (ii) extrinsic pathway, which can be initiated by ligation of death receptors followed by activation of caspase-8. Both of these pathways converge to programmed cell death that is arisen from the cleavage of caspase-7 and 3 [38–40].
Gene expression profiles in both apoptosis pathways were listed in Fig. 4. Data of extrinsic pathway suggested that the extrinsic apoptotic pathway was strongly activated in A. cantonensis-infected mice through TNFR1a and TNFR1b at week 2 PI, followed by the increased expression level of the RIPK1, FADD, and caspase-8. Furthermore, caspase-3 and 7 were also slightly up-regulated across all time points in the brain tissue, with moderately higher expression at day 2 and week 2 and 3 PI, respectively. The intrinsic pathway was consisted with anti-apoptosis mediator, Bcl-2, and downstream mediator, Bak1. Both of these 2 checkpoint genes promoted apoptosis at time point almost immediately post-infected (day 2 PI) and throughout the course of the study. Although the up-regulated Bak1 induced the discordant alternations of the downstream genes, such as cytochrome c (cyt c, in our dataset, called cycs and cyct), Apaf-1, caspase-9, that were unable to rule out use of these genes as efforts to modulate with respect to apoptosis. Thus, it suggested that neuronal apoptosis by extrinsic pathway was activated in mice in response to A. cantonensis infection.
A. cantonensis infection intensify caspase-3 expression in the cerebral cortex and hippocampus of mice
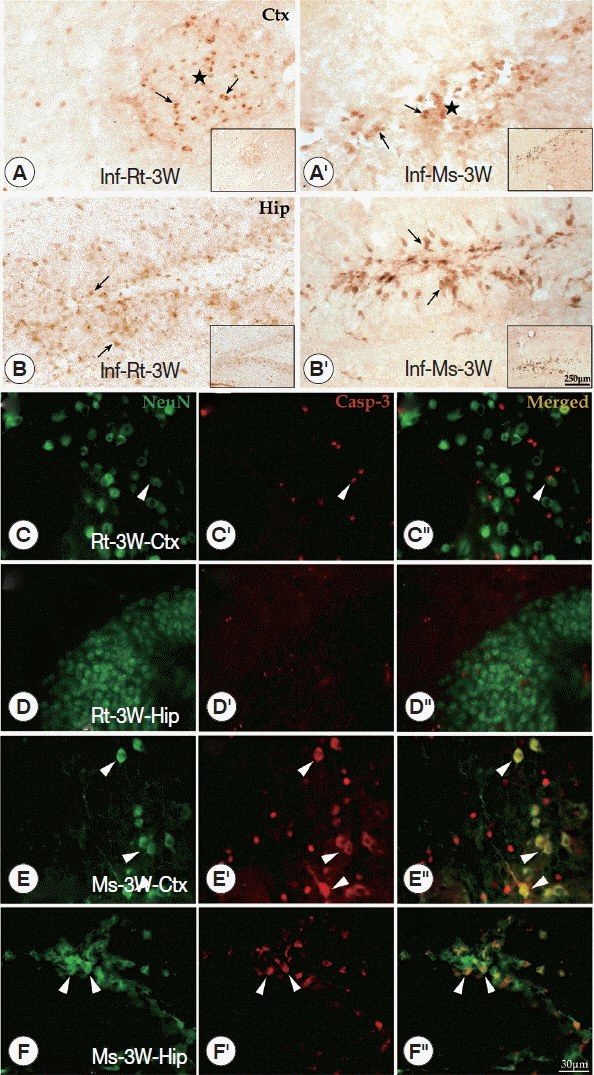
To further understand the apoptotic effects of the main pathological loci in the brain, immunohistochemistry of caspase-3 in the cerebral cortex and hippocampus was conducted. Caspase-3 positive cells (considered to be apoptotic cells) were detected at the cerebral cortex and hippocampus of both A. cantonensis-infected rodents. In the cerebral cortex, caspase-3 positive cells appeared in the center of plaque-like lesions and limited in that local region, but in the hippocampus, caspase-3 positive cells were widely visible at dentate gyrus and CA1 area (Fig. 5A-A′, B-B′). The results of double labeling for caspase-3 and NeuN showed that only a few neurons were labeled for caspase-3 at the rat cerebral cortex and hippocampus (Fig. 5C-C″, D-D″). However, numerous neurons (NeuN+) labeled for caspase-3 were detected in the mouse cerebral cortex (in plaque-like lesions) and hippocampus (especially in dentate gyrus and CA1 area) at week 3 PI (Fig. 5E-E″, F-F″). The results of both rodents showed that the density of caspase-3 positive cells of cerebral cortex and hippocampus in mice was dramatically higher than in rats at week 3 PI.
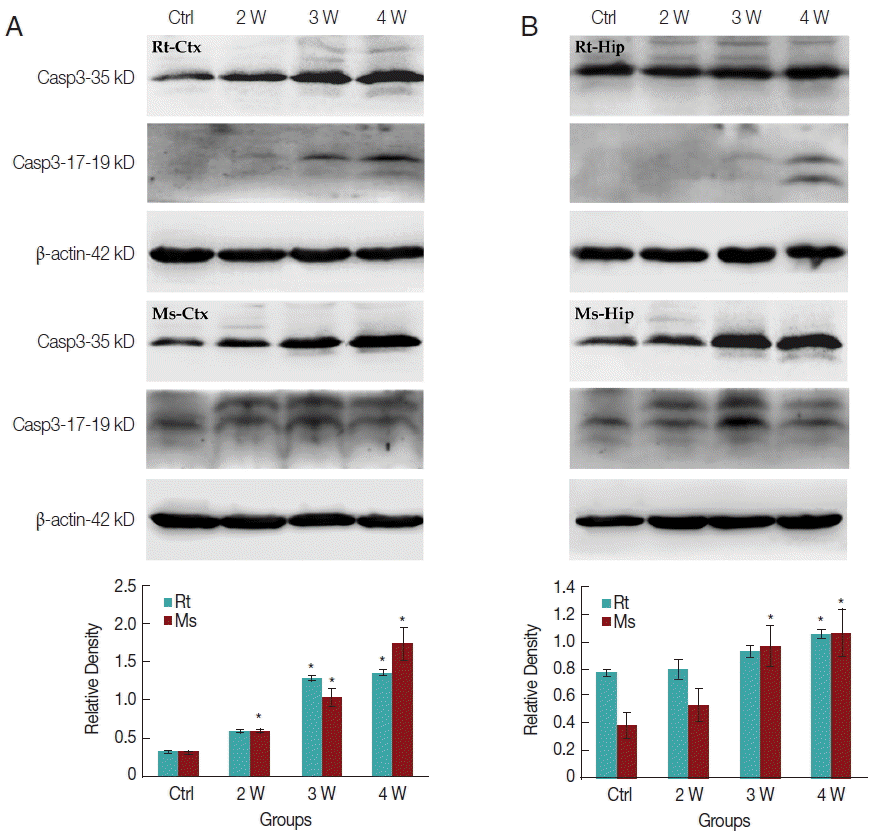
In caspase-3 dependent apoptotic pathway, the 35 kDa caspase-3 protein will be divided into 2 fragments of 17 and 19 kDa cleaved-caspase-3 protein to execute the final apoptosis. In the present study, only slightly increased caspase-3 expression in transcriptomic data could be observed. Western-blotting was further used to elucidate the expression level of caspase-3 and cleaved-caspase-3 in the cerebral cortex and hippocampus, respectively. Caspase-3 expression level was increased in the rat cerebral cortex after week 3 PI but in mice after week 2 PI (Fig. 6A). In the hippocampus, caspase-3 expression level was significantly increased in rat at week 4 PI but in mice after week 3 PI (Fig. 6B). The cleaved-caspase-3 expression level appeared slightly in the rat cerebral cortex after week 3 PI but significantly in mouse after week 2 PI (Fig. 6A). The cleaved-caspase-3 expression level also increased slightly in the rat hippocampus after week 4 PI but significantly in mouse after week 2 PI (Fig. 6B). In total, the mice cerebral cortex and hippocampus had the higher levels of caspase-3 and cleaved-caspase-3 than rats. The apoptotic effects through caspase-3 cascade pathway were significantly activated in the mice cerebral cortex and hippocampus than in the corresponding loci of rats.
DISCUSSION
Clinical exploration and animal investigation confirmed that A. cantonensis invading CNS caused the neurological symptoms with behavioral dysfunctions [10–16]. In this study, we clearly elucidated that rats and mice presented dysfunctions of movement, motor coordination, and cognition, and we further primarily explored the molecular mechanisms from the perspective of cell apoptosis by pathological observations and analysis of gene expression patterns in the apoptosis pathway. It provides reasonable explanations for the neurological symptoms in clinical patients and the behavioral disorders in animals infected with A. cantonensis.
Our previous study showed that the larvae were located within the subarachnoid space and cerebral ventricle at the early stage of infection, but in the brain parenchyma, at the late stage of infection [27]. The data from autopsies also revealed that numerous worms were presented in the subarachnoid spaces over the frontal cortex, pons, tip of the temporal lobes, and paraventricular areas of the lateral ventricle [41–43]. Detailed comparison of larvae distribution and size, and neurological damages in rats and mice with A. cantonensis infection showed that higher percentage of larvae in the rat cerebrum and higher percentage of larvae in the mice brainstem. The results further confirmed that the recovery of worms were dominant in the hippocampus of the mice, but the worms were mainly found in the cortex of the rats (Supplementary Table 7). It suggested that distinct distribution of the larvae might be related to the distinct neurological damages and symptoms in both rodents.
The major pathological damages in human CNS induced by A. cantonensis infection include; (1) meningitis with a predominance of eosinophils and plasma cells; (2) multiple microcavities, tortuous tracks, and microscopic hemorrhage surrounded by inflammatory cells and neuron lesions in the brain and spinal cord parenchyma; (3) granulomatous response to the dead worms; (4) nonspecific vascular reactions, thrombosis, and aneurysm formation [42,44]. In A. cantonensis-infected mammalian brains (permissive host rats and nonpermissive hosts, including mice, rabbits, and guinea pigs), similar pathological changes were also found in CNS [23,24,28,29,45,46]. In this study, plaque-like lesions also showed in various regions in infected animals. These regions, especially the cortex and hippocampus, execute the functions of the sense, motor, learning, and memory. The histopathologic investigations also indicated that the plaque-like lesions were infiltrated by numerous eosinophils, with neurons lost in the lesion core in the cortex (sense and motor) and hippocampus (learning and memory). Thus, it is suggested that the worm distribution caused the damages in the corresponding regions, leading to functional impairments. Similar results have been reported in Toxoplasma gondii infected mice which revealed reduced exploratory activity according to the climbing and rearing, showing more exposed part of the OF arena, smaller preference for the central, and less grooming behavior, which were related to the presence of cysts both in the amygdala and hippocampus [47]. Another study showed that a high density of lesions, such as the recruited immune cells and the presence of activated resident glia accompanied by limited alterations of neuronal connectivity abnormalities, was observed within the somatosensory cortex (SSC). Altered fiber density indicated a reduction in spine density and dendritic arbor in the SSC and hippocampus [48]. Moreover, a previous study demonstrated that double immunofluorescent staining of NeuN and caspase-3 resulted into the neuronal apoptosis during T. gondii infection [49]. Therefore, the investigations in this study of immunohistochemical staining and double-labeling for NeuN and caspase-3 showed that numerous apoptotic neurons were widely distributed in the hippocampal dentate gyrus and CA1 compared to the nidus limited in a local region in the cortex, which might be one of the explanations for behavioral dysfunctions in animals and neurological symptoms in clinical patients.
Memory loss and cognitive impairment are always closely correlated with neuronal apoptosis. In our study, the dramatically increased TNFR1 and caspase-8 expression in the brain tissue was observed in mice with A. cantonensis infection. In the extrinsic pathway, TNFR1 plays an important role by recruiting the cytosolic adaptor FADD and RIPK1, which are associated with caspase-8 in a variety of immune and parenchymal tissues [50]. Furthermore, it is TNFR1 that induces extrinsic apoptosis through RIPK1 and caspase-8 in Yersinia-induced cell death of murine macrophages [51]. Furthermore, cleaved-caspase-3 expression level was significantly increased in the cortex and hippocampus in mice but not in rats, indicating that neuronal apoptosis by TNFR1 and caspase-8 pathway might be the key molecular mechanism to induce the neuronal injury with A. cantonensis infection. These results were consistent to the distinct extent of the behavioral dysfunction in mice and rats.
In summary, all these recovery rate, pathologic evidences in the cerebral cortex and hippocampus, and activated extracellular caspase-dependent apoptosis pathway in mice and rats indicated that mice (nonpermissive host) experienced more serious pathological injuries with the caspase-dependent neuronal cell apoptosis than rats (permissive host). The neurological symptoms in clinical infected patients can be explained using the behavioral disorders in our experimental model. The results of mice with A. cantonensis infection will be important for clinical diagnosis and therapy. It also provides evidence that neuroprotection should be considered for the patients with A. cantonensis infection.